
Hematology — MCQs

On this page
Spherocytes are not seen in which of the following conditions?
A 15-year-old boy presents with acute onset of right flank pain that developed after physical exercise. Physical examination demonstrates an area of ecchymosis in the right flank that is tender to palpation. The patient has a lifelong history of easy bruising, and a sibling has the same symptoms. The serum level of clotting factor VIII is less than 2% of normal. Which of the following is the most likely underlying mechanism for the bleeding tendency in this patient?
A 48-year-old woman presents with a 3-week history of fatigue and yellow skin and sclerae. Physical examination reveals mild jaundice. Laboratory findings include a serum bilirubin level of 3.7 mg/dL (predominantly unconjugated), normal serum AST, ALT, and alkaline phosphatase, and a hemoglobin level of 6.0 g/dL. Jaundice resolved after administration of corticosteroids. What is the most likely cause of hyperbilirubinemia in this patient?
Which drug is associated with warm antibody hemolytic anemia?
A 46-year-old woman with prominent splenomegaly presents with a 3-month history of malaise, easy fatigability, weakness, weight loss, and anorexia. A complete blood count and differential demonstrates a white blood cell count of 250,000/mm3 with a predominance of myelocytes, metamyelocytes, band cells, and segmented neutrophils. Cytogenetic analysis is most likely to reveal which of the following translocations?
Which of the following is NOT true of long-standing sickle cell anemia?
Which of the following is NOT a feature of Antiphospholipid Syndrome (APS)?
A 69-year-old woman presents with fatigue. Her hemoglobin is 9.0 g/dL (14 g/dL one year prior) with no other symptoms. A blood film is provided. What is the most appropriate next diagnostic step?
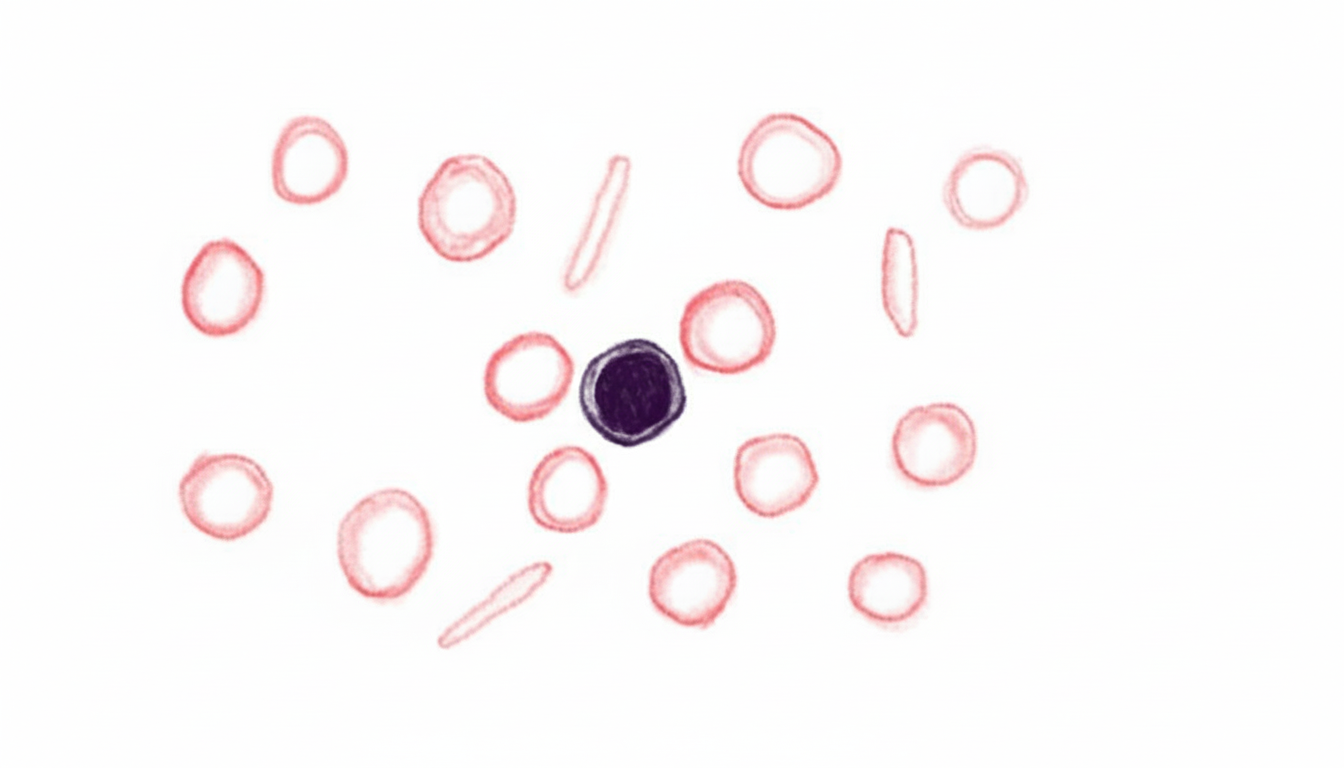
To which of the following diseases is pyruvate kinase deficiency most similar clinically?
Which of the following is a characteristic feature of hypoproliferative anemia?
Practice by Chapter
Anemia Evaluation and Management
Practice Questions
Hemoglobinopathies
Practice Questions
Thalassemias
Practice Questions
Platelet Disorders
Practice Questions
Coagulation Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Leukemias
Practice Questions
Lymphomas
Practice Questions
Multiple Myeloma and Plasma Cell Disorders
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Transfusion Medicine
Practice Questions
Hematopoietic Stem Cell Transplantation
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app