
Hematology — MCQs

On this page
A 45-year-old female patient presents with symptoms of easy bruisability and frequent headaches. Physical examination shows moderate splenomegaly. Blood counts show a normal leucocyte count and a platelet count of 1000 × 10^3/cu mm. The leucocyte alkaline phosphatase score is normal. Which one of the following is the drug of choice for the treatment of this patient?
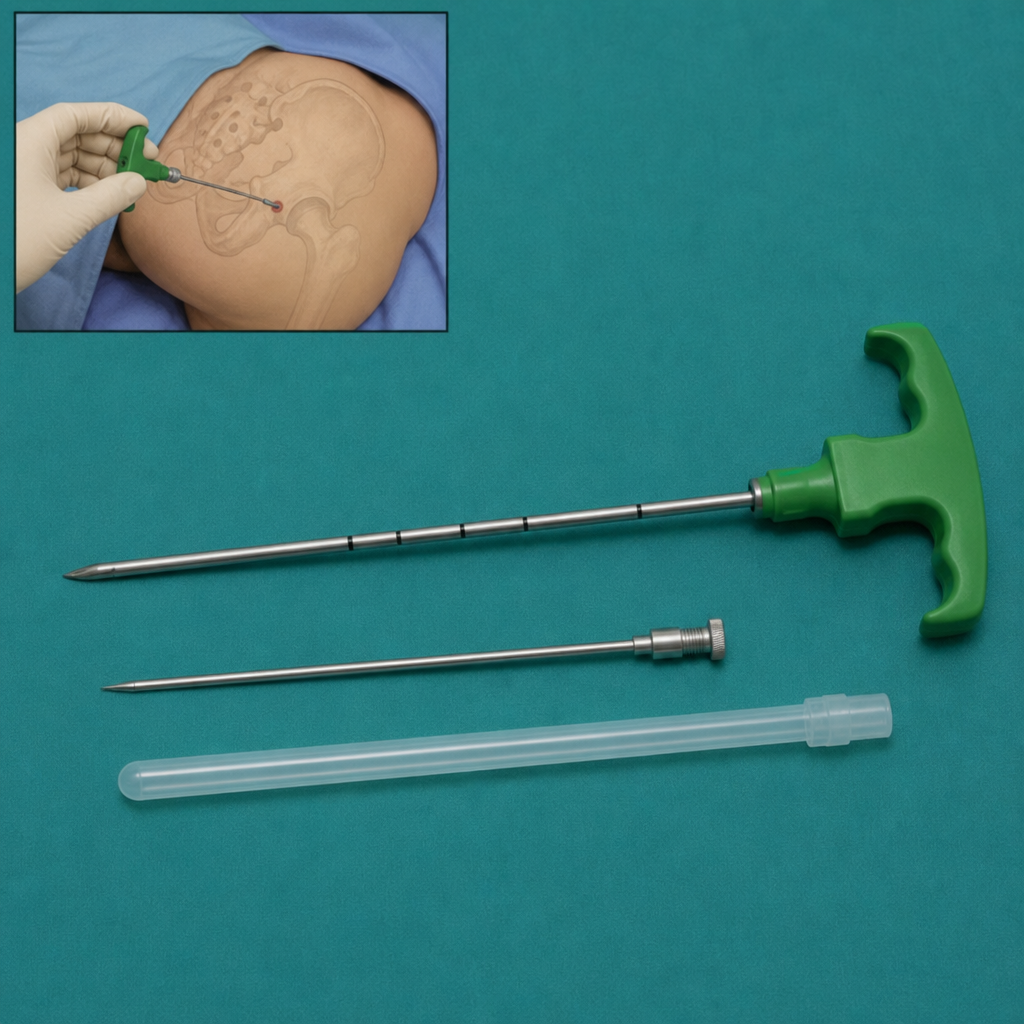
For what procedure is the instrument depicted in the diagram used?
Which condition is associated with Coomb's positive hemolytic anemia?
Elevated serum ferritin, decreased serum iron, and decreased % transferrin saturation are most consistent with which diagnosis?
A 30-year-old male, undergoing platelet apheresis for the first time, experiences tingling around the mouth and numbness in his arm. An ECG shows ST segment elevation. What is the most likely cause of these symptoms?
A 40-year-old chronic alcoholic is investigated for anemia. Tests reveal increased serum iron and increased transferrin saturation. What is the probable diagnosis?
All of the following are conditions that precipitate hemolysis in G6PD deficiency, except?
What is the positive finding in a patient with anemia due to chronic inflammation?
Hunter's glossitis is a feature of which condition?
A 65-year-old man presents with tingling in his hands and feet and increased forgetfulness. A CBC shows mild anemia. The patient reports no change in diet, other than increased consumption of red meat. What is the most appropriate treatment for this patient?
Practice by Chapter
Anemia Evaluation and Management
Practice Questions
Hemoglobinopathies
Practice Questions
Thalassemias
Practice Questions
Platelet Disorders
Practice Questions
Coagulation Disorders
Practice Questions
Thrombotic Disorders
Practice Questions
Leukemias
Practice Questions
Lymphomas
Practice Questions
Multiple Myeloma and Plasma Cell Disorders
Practice Questions
Myeloproliferative Neoplasms
Practice Questions
Transfusion Medicine
Practice Questions
Hematopoietic Stem Cell Transplantation
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app