
Endocrinology — MCQs

On this page
A 60-year-old male presented with skin hyperpigmentation, central obesity, violet striae, and proximal myopathy. His blood pressure is 160/90 mmHg. Lab studies show hypokalemic metabolic alkalosis. Which of the following statements is true regarding this condition?
Chvostek sign is seen in:
Vitamin D resistant rickets occurs due to all EXCEPT:
A smoker presented with altered sensorium. His blood osmolality was found to be 240 mOsm/kg, urine osmolality 340 mOsm/kg, and serum Na+ is 122 mEq/L. What is the most likely diagnosis?
Which vaccine can be given to an individual with chronic diabetes mellitus?
Hypothyroidism is seen in which of the following conditions?
A 33-year-old woman presents with gradually increasing coarse hair on her upper lip, chin, and lower abdomen for the past 3 years. She has mild facial acne but denies frontal balding or deepening of voice. Her menses are irregular, occurring every 28 to 60 days. Her BMI is 29.0 and her waist circumference is 36 inches. There is mild acanthosis nigricans of the axillae. Pelvic examination is normal without ovarian mass or clitoromegaly. What evaluation is indicated for her hirsutism?
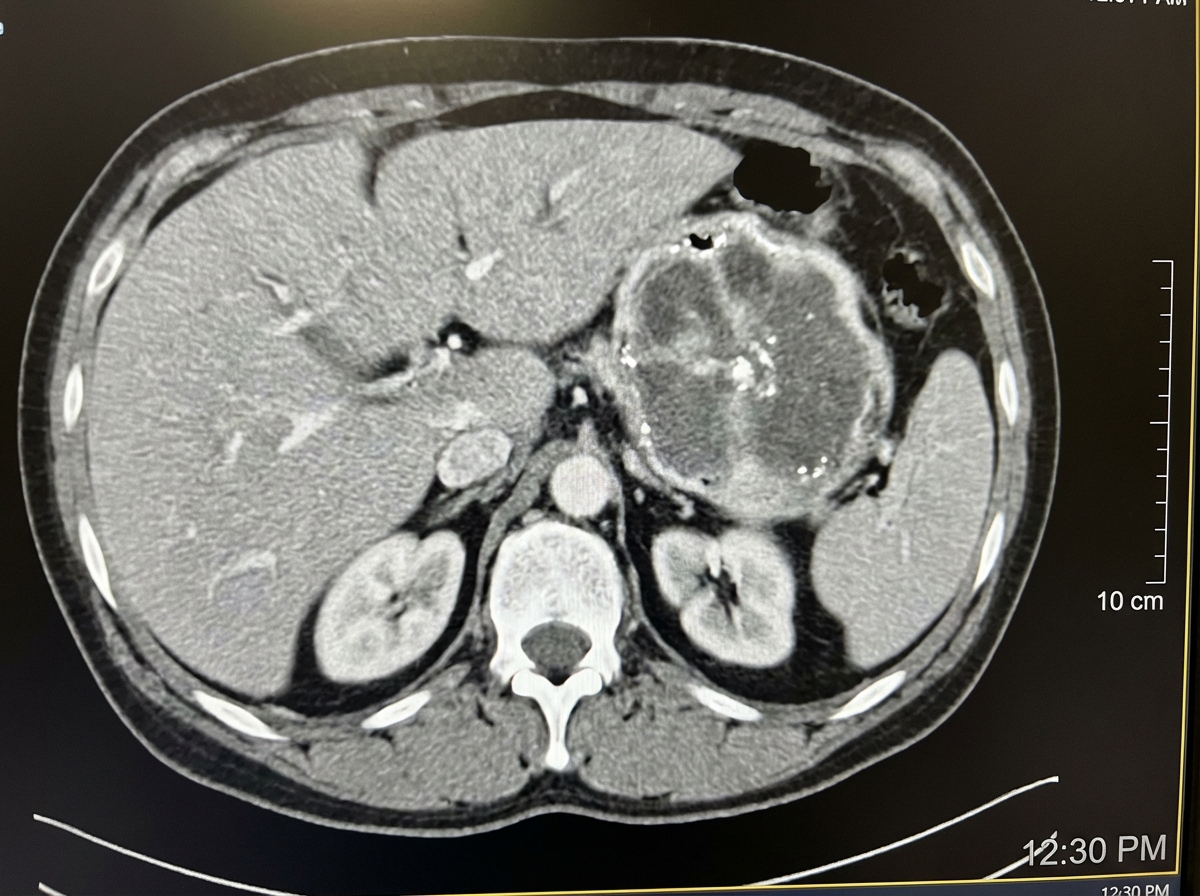
A 40-year-old patient presents with episodic hypertension, severe headache, diaphoresis, and palpitations. The CT scan of the abdomen is shown in the image. What is the most likely diagnosis?
A 18-year-old male presents with severe weakness, dizziness, sleepiness, unquenchable thirst, and increased urination over the past 4 weeks. He has also experienced drowsiness and generalized tiredness in a previous episode 3 weeks earlier and has lost 4kg of weight. Laboratory findings on admission show a glucose of 560 mg/dl, urine that is 4+ for glucose and has "large" acetone, and an HbA1c of 14%. Which of the following is true regarding this patient?
What is the primary medical treatment for hyperprolactinemia?
Practice by Chapter
Diabetes Mellitus
Practice Questions
Thyroid Disorders
Practice Questions
Adrenal Gland Disorders
Practice Questions
Pituitary Disorders
Practice Questions
Calcium and Bone Metabolism
Practice Questions
Reproductive Endocrinology
Practice Questions
Lipid Disorders
Practice Questions
Endocrine Hypertension
Practice Questions
Multiple Endocrine Neoplasia
Practice Questions
Obesity and Metabolic Syndrome
Practice Questions
Neuroendocrine Tumors
Practice Questions
Endocrine Emergencies
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app