
Pediatric Dermatology — MCQs

On this page
What are the common sites for a Mongolian spot?
Koenen's periungual fibromas are seen in > 50% of cases with which condition?
A patient presents with seizures, adenoma sebaceum, and mental retardation. What is the diagnosis?
What is the recommended treatment for a child presenting with an erythematous, non-blanching, bosselated lesion on the right side of the face?
A 1 and 1/2 year old child with a history of chronic diarrhea presents with perioral and perineal rash. What is your diagnosis?
What is the best treatment for a strawberry angioma?
Which of the following is NOT true regarding tuberous sclerosis?
Sturge-Weber syndrome is associated with which of the following?
Which of the following conditions is characterized by the finding shown below?
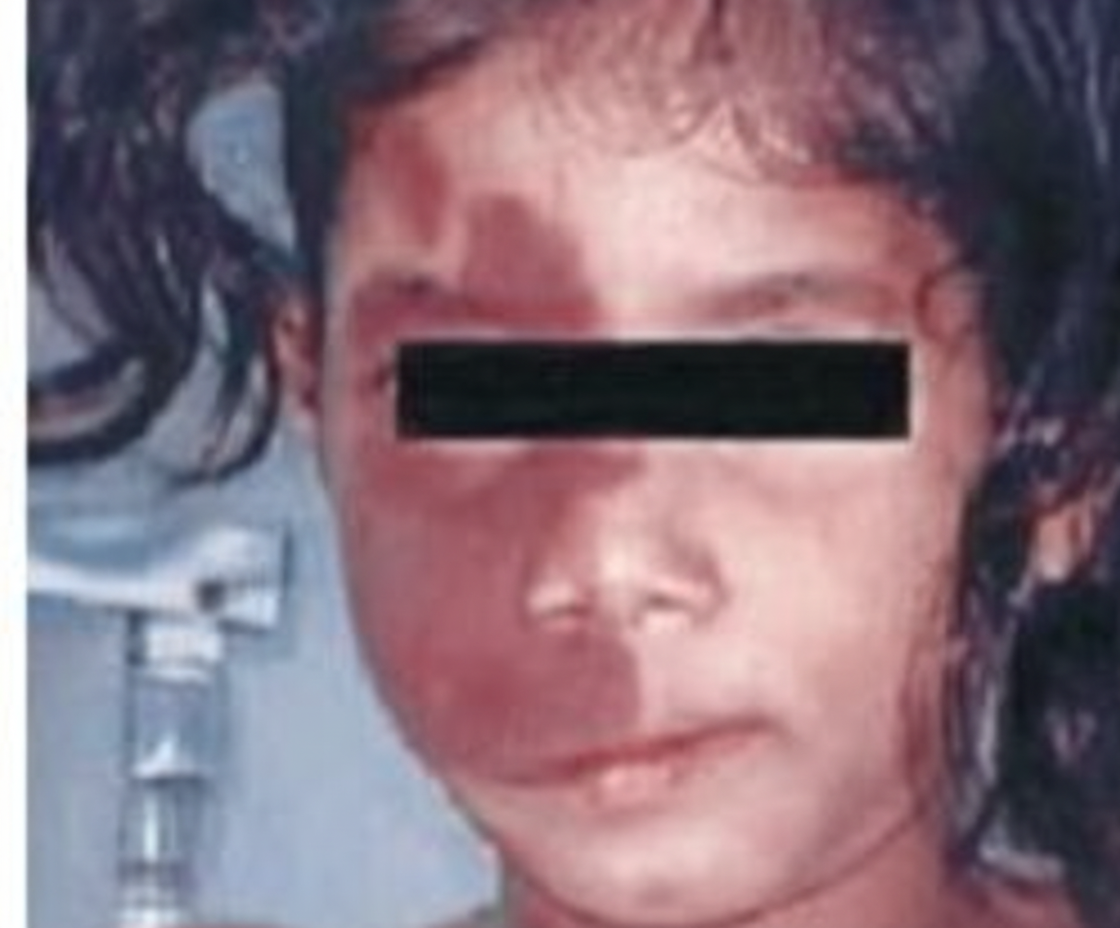
What is the most common clinical sign or symptom for the diagnosis of PHACE syndrome?
Practice by Chapter
Neonatal Dermatology
Practice Questions
Infantile Hemangiomas and Vascular Malformations
Practice Questions
Atopic Dermatitis in Children
Practice Questions
Acne in Childhood and Adolescence
Practice Questions
Childhood Exanthems
Practice Questions
Genetic Skin Disorders in Children
Practice Questions
Genodermatoses
Practice Questions
Nutritional Dermatoses in Children
Practice Questions
Pigmentary Disorders in Children
Practice Questions
Hair Disorders in Children
Practice Questions
Child Abuse: Cutaneous Manifestations
Practice Questions
Therapeutic Considerations in Pediatric Dermatology
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app