
Pediatric Dermatology — MCQs

On this page
Which of the following is NOT a feature of Neurofibromatosis-1?
A 6-year-old child is brought by his new foster mother who was concerned that when she brushed his teeth last night she noticed that his tongue was red in certain distinct patterns. He has otherwise not been ill. What is the most likely diagnosis?
A 17-year-old pregnant woman presents with scattered small, raised lesions on her trunk and axillary freckles. What is the most likely mode of inheritance for this condition?
A child is diagnosed to have tuberous sclerosis. Which of the following skin lesions is not associated?
In addition to neurofibromatosis, what other examination finding would you expect for this patient?
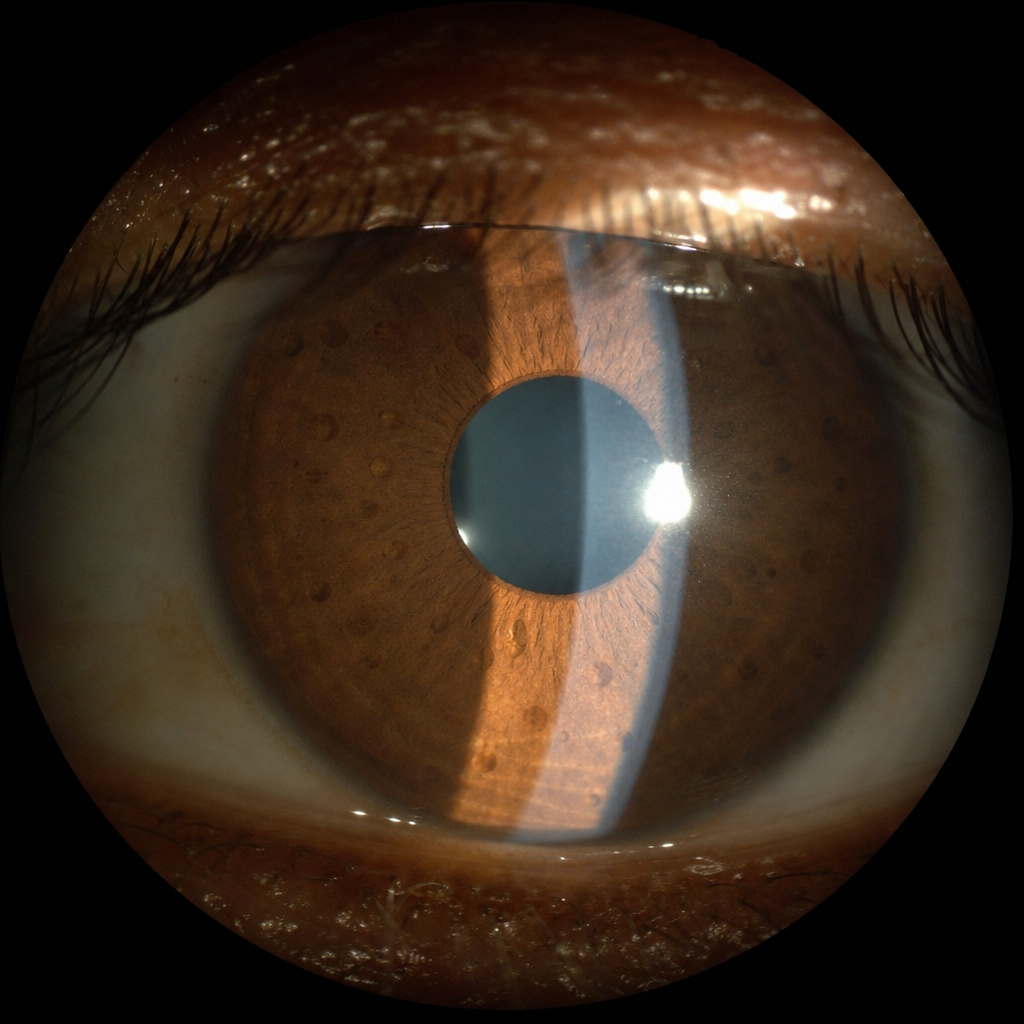
Which condition is characterized by the following lesion?
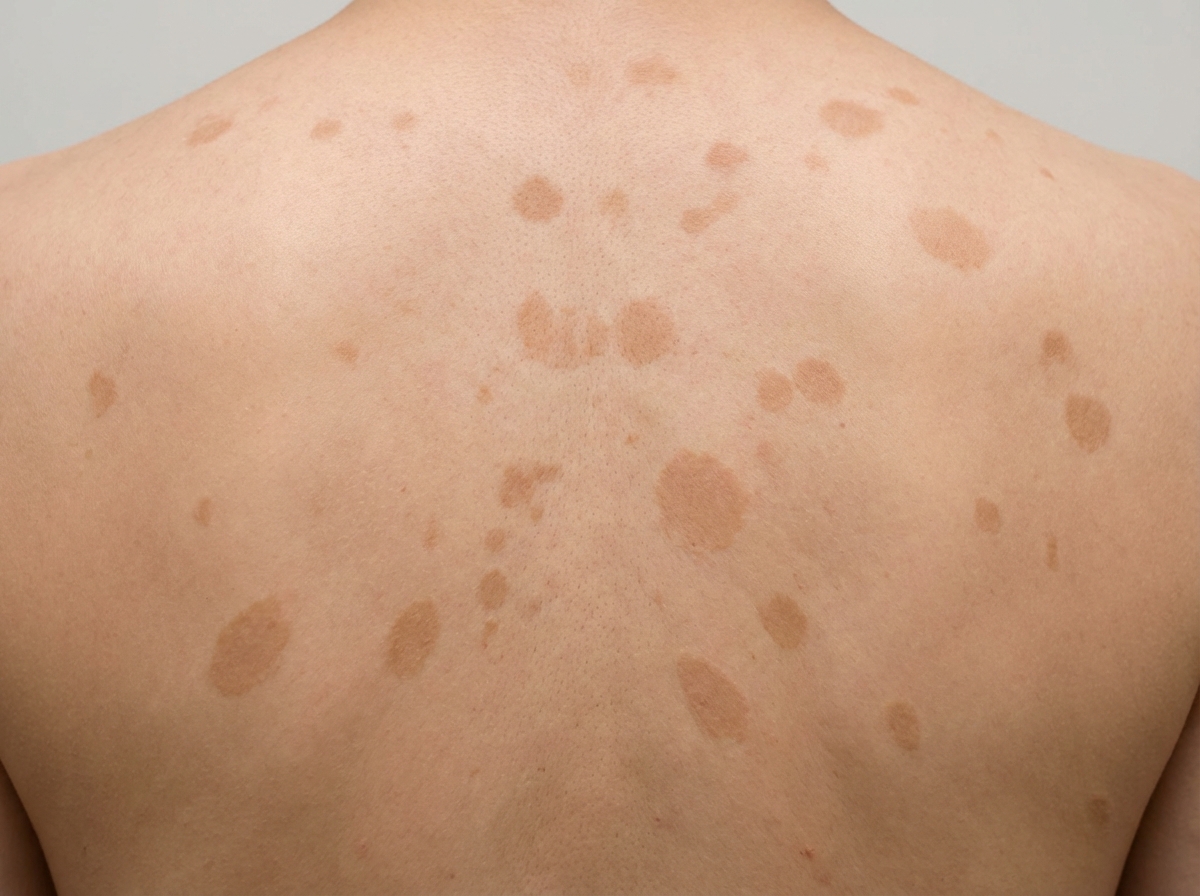
Spontaneous regression can occur with which of the following conditions?
A 10-year-old child has violaceous papules and pterygium of the nails. What is the most likely diagnosis?
An asymptomatic 3-year-old child presents with acute onset right cheek swelling that is erythematous but not warm to touch. On palpation, mildly tender, discrete, indurated masses are appreciated. What is the most likely cause of this presentation?
Neonatal fat necrosis (subcutaneous fat necrosis of the newborn) resembles which of the following conditions?
Practice by Chapter
Neonatal Dermatology
Practice Questions
Infantile Hemangiomas and Vascular Malformations
Practice Questions
Atopic Dermatitis in Children
Practice Questions
Acne in Childhood and Adolescence
Practice Questions
Childhood Exanthems
Practice Questions
Genetic Skin Disorders in Children
Practice Questions
Genodermatoses
Practice Questions
Nutritional Dermatoses in Children
Practice Questions
Pigmentary Disorders in Children
Practice Questions
Hair Disorders in Children
Practice Questions
Child Abuse: Cutaneous Manifestations
Practice Questions
Therapeutic Considerations in Pediatric Dermatology
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app