
Hemoglobin and Iron Metabolism — MCQs

On this page
Iron absorption is increased in which of the following conditions?
What is the end product of porphyrin metabolism?
The affinity of hemoglobin for O2 is increased by:
Conjugated hyperbilirubinemia is seen in which of the following conditions?
Patients with axonal neuropathy, unexplained abdominal pain, and a history of psychiatric illness may be suffering from which condition?
What is the composition of HbA2?
Which of the following statements is NOT true regarding iron metabolism?
Which of the following statements regarding heme synthesis is FALSE?
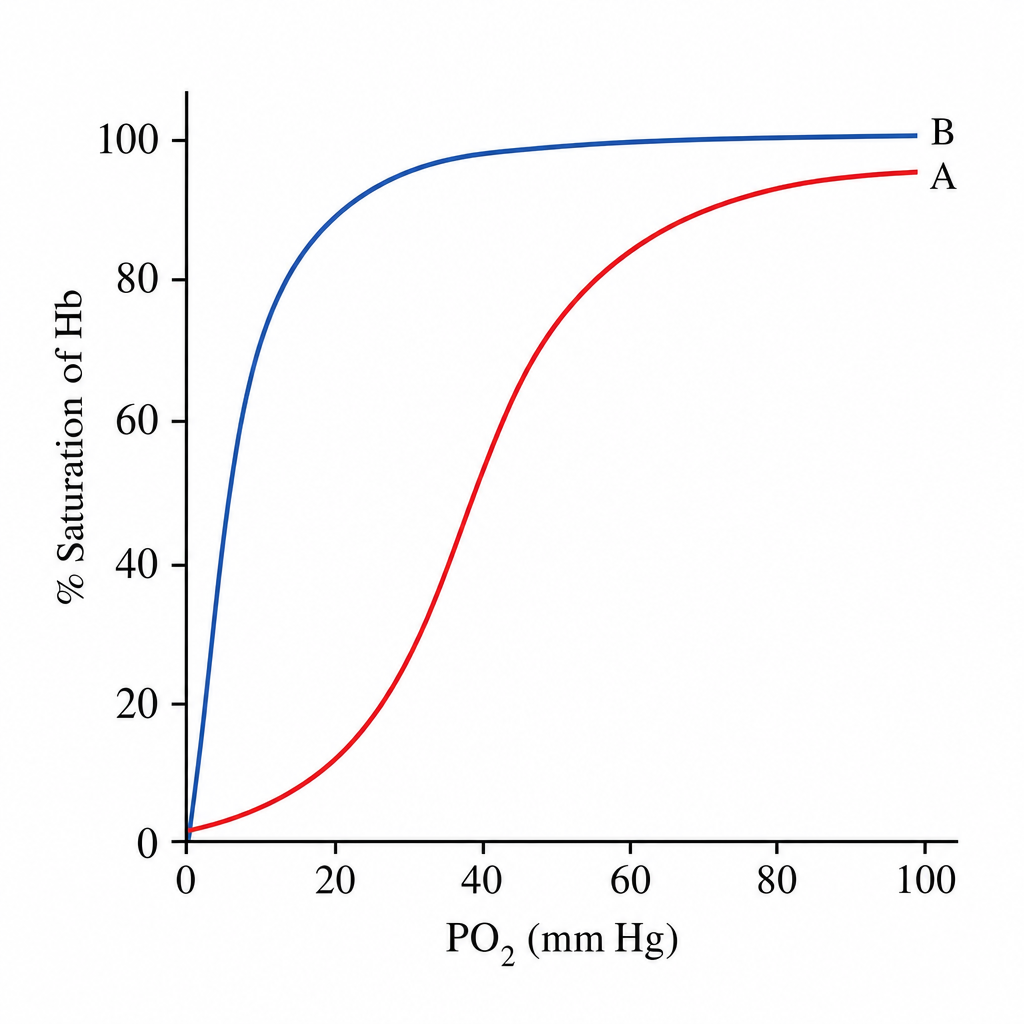
Study the given curve and comment on the finding.
In chronic lead poisoning, which of the following is elevated?
Practice by Chapter
Hemoglobin Structure and Function
Practice Questions
Oxygen Transport and Oxygen-Hemoglobin Dissociation Curve
Practice Questions
Hemoglobin Variants and Hemoglobinopathies
Practice Questions
Thalassemias
Practice Questions
Methemoglobin and Abnormal Hemoglobins
Practice Questions
Hemoglobin Synthesis
Practice Questions
Heme Synthesis and Porphyrias
Practice Questions
Iron Absorption and Transport
Practice Questions
Iron Storage and Recycling
Practice Questions
Disorders of Iron Metabolism
Practice Questions
Anemia: Biochemical Aspects
Practice Questions
Biochemistry of Hemostasis
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app