
Hemoglobin and Iron Metabolism — MCQs

On this page
What enzyme is deficient in erythropoietic porphyria?
Plasma ferritin levels may be reduced in all of the following conditions, except?
Which of the following statements about bilirubin is false?
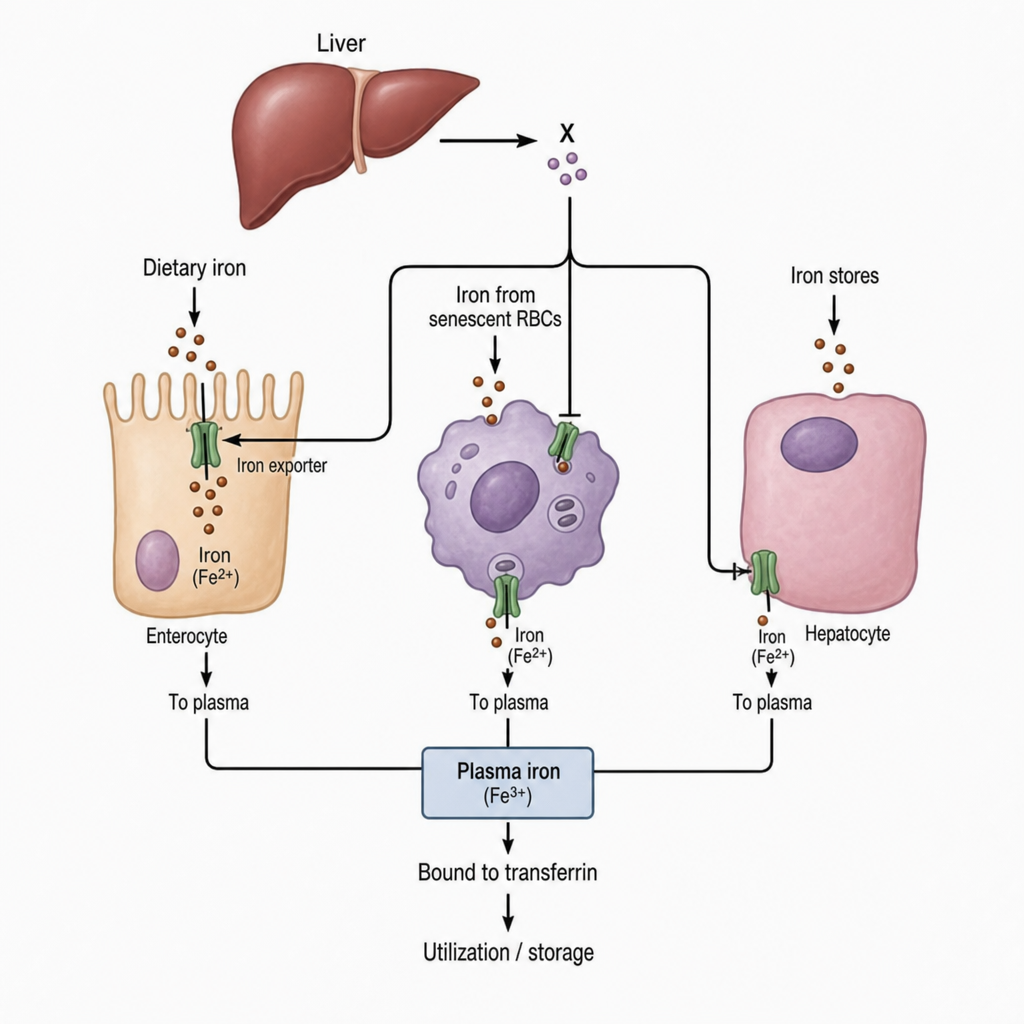
Which of the following proteins involved in iron metabolism denotes X?
Which of the following facilitates the absorption of iron from the intestine?
High affinity of HbF with O2 is due to:
In which of the following laboratory tests would you expect to find the greatest disparity in reference intervals between men and non-pregnant women?
Which of the following conditions is suggested by elevated serum ferritin, serum iron, and percent transferrin saturation?
Which among the following is the major hemoglobin found in the fetus?
Which one of the following amino acids in Hemoglobin accepts H+ and allows Hemoglobin to act as a buffer to acids?
Practice by Chapter
Hemoglobin Structure and Function
Practice Questions
Oxygen Transport and Oxygen-Hemoglobin Dissociation Curve
Practice Questions
Hemoglobin Variants and Hemoglobinopathies
Practice Questions
Thalassemias
Practice Questions
Methemoglobin and Abnormal Hemoglobins
Practice Questions
Hemoglobin Synthesis
Practice Questions
Heme Synthesis and Porphyrias
Practice Questions
Iron Absorption and Transport
Practice Questions
Iron Storage and Recycling
Practice Questions
Disorders of Iron Metabolism
Practice Questions
Anemia: Biochemical Aspects
Practice Questions
Biochemistry of Hemostasis
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app