
Hemoglobin and Iron Metabolism — MCQs

On this page
Hemoglobin is present in which type of pockets?
Hemoglobin does not bind with:
The heme portion of the hemoglobin molecule consists of:
Along with succinyl CoA, which of the following amino acids serves as a starting material in heme synthesis?
A 34-year-old female has a history of intermittent episodes of severe abdominal pain. She has had multiple abdominal surgeries and exploratory procedures with no abnormal findings. Her urine appears dark during an attack and gets even darker if exposed to sunlight. The attacks seem to peak after she takes erythromycin, because of her penicillin allergy. This patient most likely has difficulty in synthesizing which one of the following?
Iron absorption is decreased in which of the following?
A patient who was in excruciating pain all over his body was taken to the hospital. In recent years, he has experienced these episodes frequently. When exercising vigorously, the pain begins. Anemia was detected on blood examination along with sickled RBCs as opposed to normal biconcave ones. It was determined that he had sickle cell anemia. What substitution takes place in sickle cell anemia?
Which of the following correctly represents the effect of the mutation causing sickle cell anemia?
Hemoglobin with iron in ferric form is
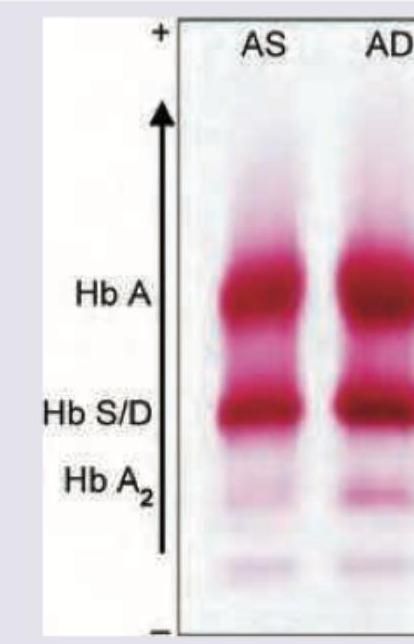
The shown pattern in electrophoresis is due to:
Practice by Chapter
Hemoglobin Structure and Function
Practice Questions
Oxygen Transport and Oxygen-Hemoglobin Dissociation Curve
Practice Questions
Hemoglobin Variants and Hemoglobinopathies
Practice Questions
Thalassemias
Practice Questions
Methemoglobin and Abnormal Hemoglobins
Practice Questions
Hemoglobin Synthesis
Practice Questions
Heme Synthesis and Porphyrias
Practice Questions
Iron Absorption and Transport
Practice Questions
Iron Storage and Recycling
Practice Questions
Disorders of Iron Metabolism
Practice Questions
Anemia: Biochemical Aspects
Practice Questions
Biochemistry of Hemostasis
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app