
Hemoglobin and Iron Metabolism — MCQs

On this page
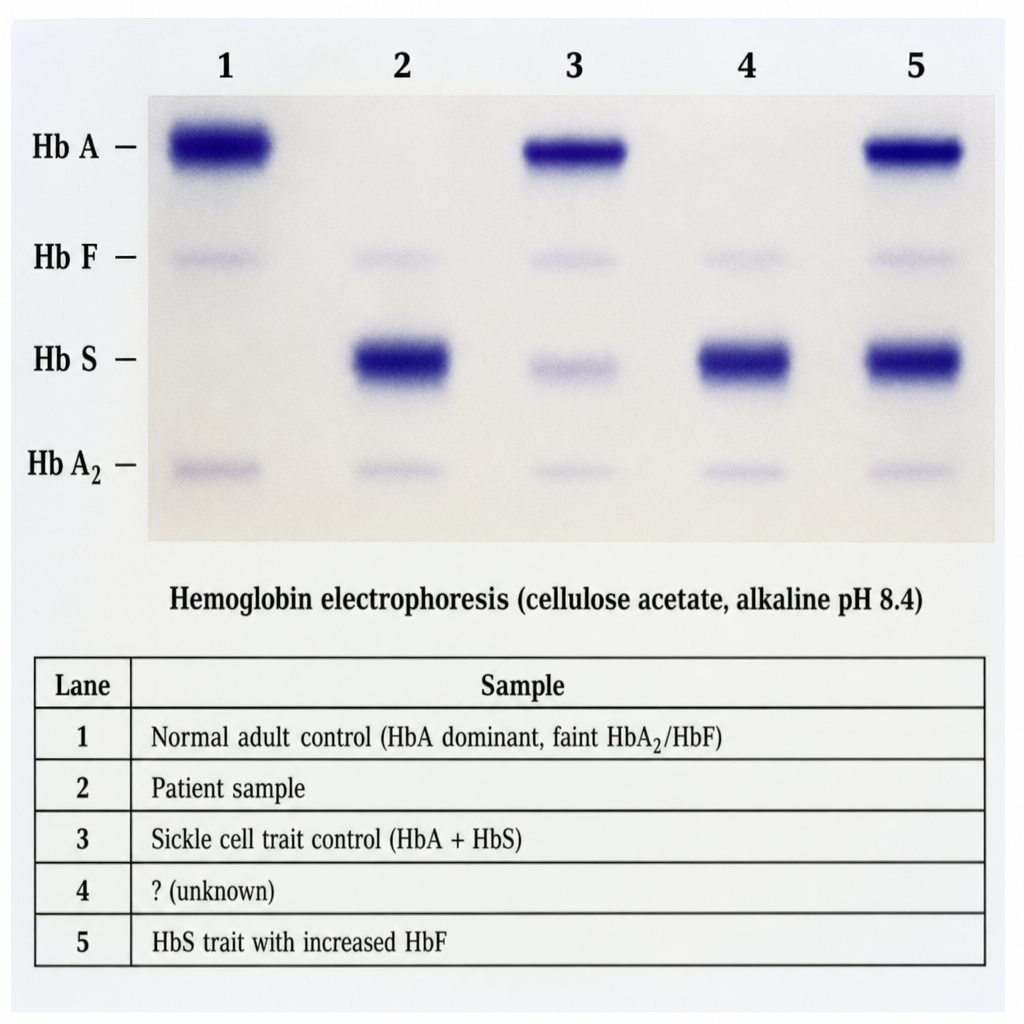
A 19-year-old male of West African descent presents with recurrent episodes of bone pain, acute chest syndrome, and a haemoglobin of 8.2 g/dL with target cells and sickle forms on peripheral smear. His parents are both clinically well. Haemoglobin electrophoresis (cellulose acetate, alkaline pH 8.4) is performed and shown in Image 1. The five lanes are: Lane 1 = Normal adult control (HbA dominant, faint HbA2/HbF); Lane 2 = Patient sample; Lane 3 = Sickle cell trait control (HbA + HbS); Lane 4 = HbC disease control (HbC only); Lane 5 = HbSC disease control (HbS + HbC, roughly equal intensity). Reference positions for HbA, HbS, HbC, and HbA2 are labeled on the gel. Lane 2 shows two bands — one co-migrating with HbS and one co-migrating with HbC — with complete absence of HbA. Based on the electrophoresis pattern in Lane 2, which combination of amino acid substitutions in the beta-globin chain is responsible for the two abnormal haemoglobin variants seen in this patient?
Ferritin biosynthesis is regulated by the serum level of which substance?
Which of the following is required in the initial stage of the synthesis of haemoglobin?
Which enzyme is involved in the enzymatic defect of Variegate porphyria?
Oral contraceptive pills (OCPs) intake can cause psychiatric symptoms and abdominal pain. What is the most likely diagnosis associated with these symptoms?
A defect in the MRP2 transporter leads to which of the following conditions?
Which of the following is considered embryonic hemoglobin?
Which of the following decreases the absorption of iron from the intestine?
Which micronutrient deficiency causes anemia?
Red cell protoporphyrin levels more than 100 micrograms/dL are suggestive of which of the following conditions?
Practice by Chapter
Hemoglobin Structure and Function
Practice Questions
Oxygen Transport and Oxygen-Hemoglobin Dissociation Curve
Practice Questions
Hemoglobin Variants and Hemoglobinopathies
Practice Questions
Thalassemias
Practice Questions
Methemoglobin and Abnormal Hemoglobins
Practice Questions
Hemoglobin Synthesis
Practice Questions
Heme Synthesis and Porphyrias
Practice Questions
Iron Absorption and Transport
Practice Questions
Iron Storage and Recycling
Practice Questions
Disorders of Iron Metabolism
Practice Questions
Anemia: Biochemical Aspects
Practice Questions
Biochemistry of Hemostasis
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app