
Genetic Disorders and Biochemical Pathology — MCQs

On this page
Inheritance associated with congenital adrenal hyperplasia -
What is the most definitive method for confirming 46,XY disorders of sexual development?
Which of the following statements is true regarding hemophilia inheritance?
Which type of genetic disorders are more likely to affect boys?
Pendred syndrome is caused by a mutation in which gene?
Gene responsible for Wilson disease is situated on which chromosome?
An infant is brought by his parents with complaints that his urine turns black on standing. Which of the following metabolic disorders is likely?
In argininosuccinase deficiency, what should be supplemented to continue the urea cycle ?
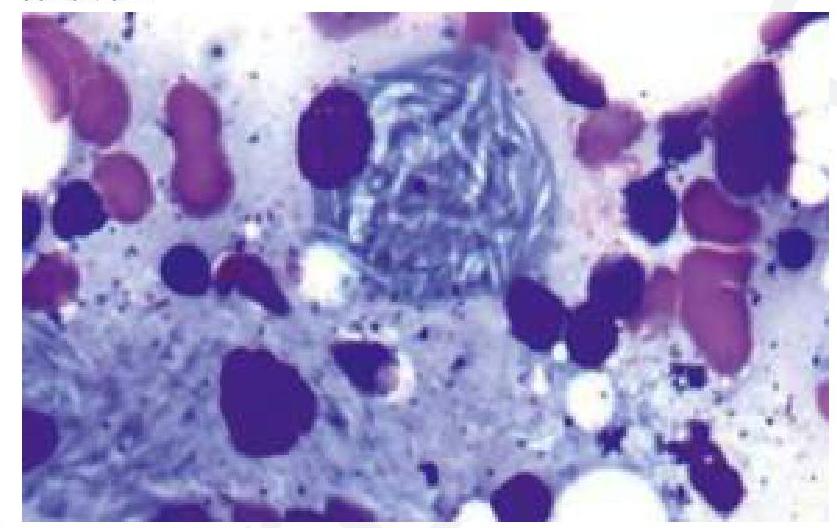
A child presents with bone pain and hepatosplenomegaly, indicative of Gaucher's disease. A trephine biopsy and aspirate show the following finding. Which of the following is the most likely enzyme deficient in this condition?
Werner syndrome associated with premature aging is caused due to a defect in which of the following?
Practice by Chapter
Single Gene Disorders
Practice Questions
Biochemical Diagnosis of Genetic Disorders
Practice Questions
Inborn Errors of Metabolism
Practice Questions
Lysosomal Storage Diseases
Practice Questions
Glycogen Storage Diseases
Practice Questions
Disorders of Lipoprotein Metabolism
Practice Questions
Disorders of Purine and Pyrimidine Metabolism
Practice Questions
Hemoglobinopathies
Practice Questions
Porphyrias
Practice Questions
Biochemical Markers for Disease Diagnosis
Practice Questions
Newborn Screening for Genetic Disorders
Practice Questions
Enzyme Replacement Therapy
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app