
Genetic Disorders and Biochemical Pathology — MCQs

On this page
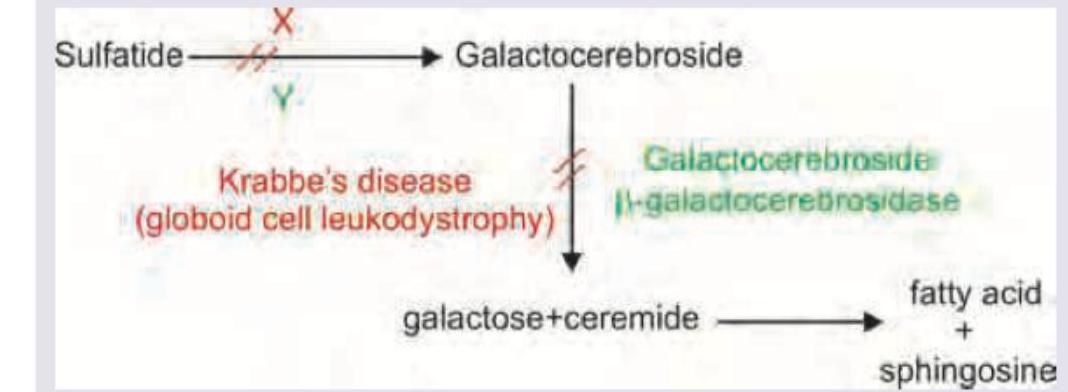
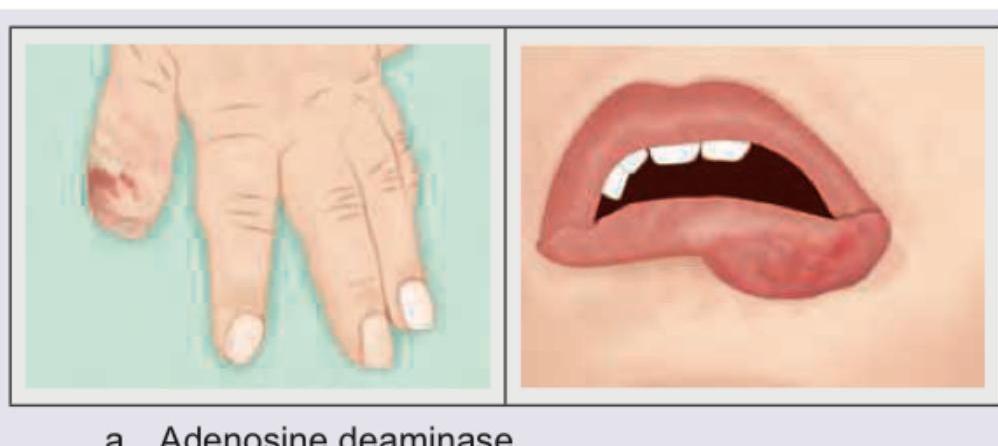
Which of the following is correct about the disease and its missing enzyme mentioned below?
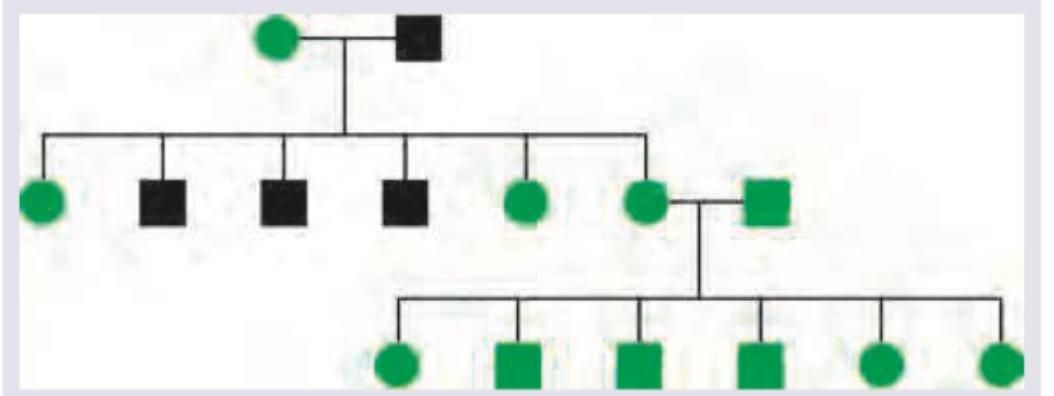
Identify the pattern of inheritance shown below.
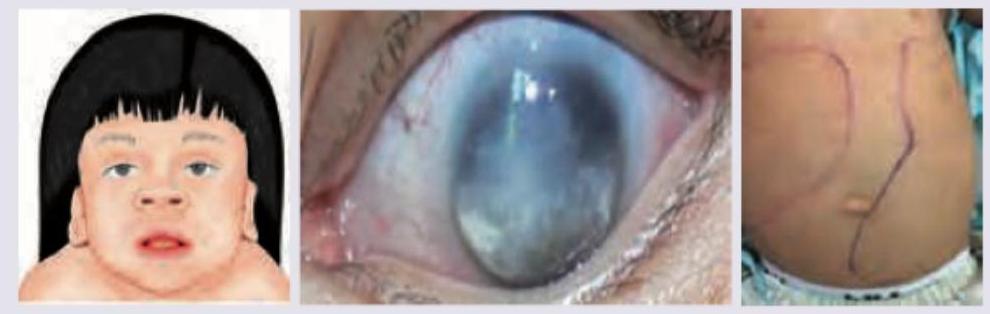
During evaluation of a child with intellectual disability following findings were noted. These point to deficiency of?
A floppy infant was brought with difficulty in breathing. CXR was performed. Family history was positive for death of sibling at 1-year of age. Which of the following enzyme deficiency will lead to this presentation?
A patient reports a change in colour of urine on air exposure. All are true about the condition shown below except:
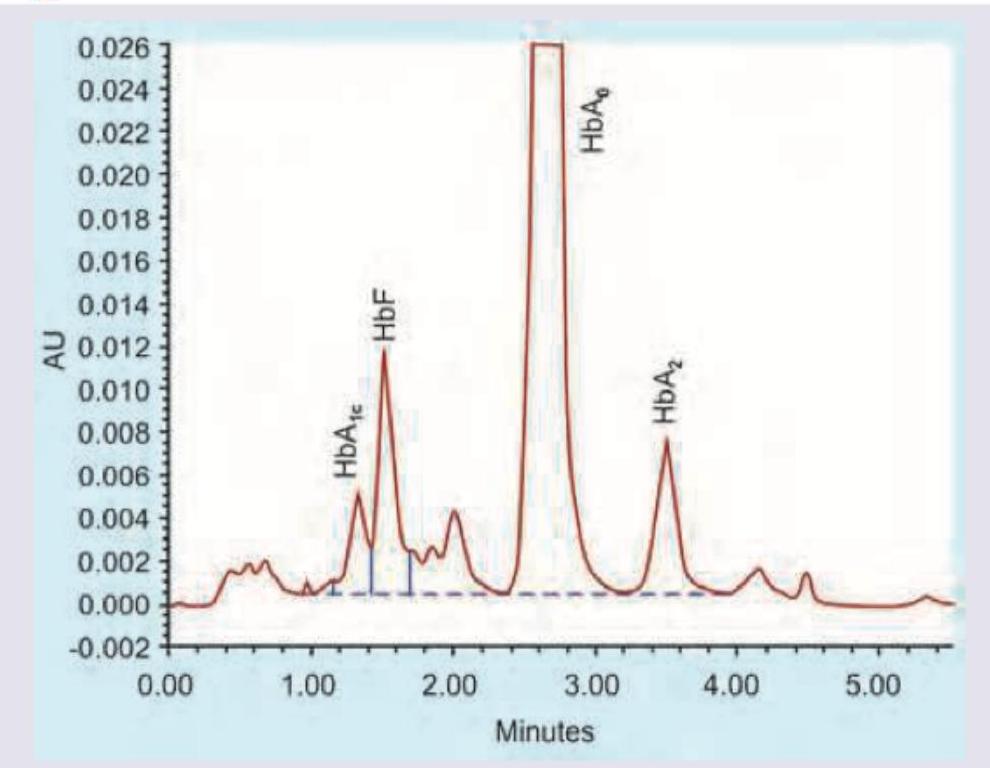
The technique shown in the image is:
The patient shown here is suffering from deficiency of which enzyme?
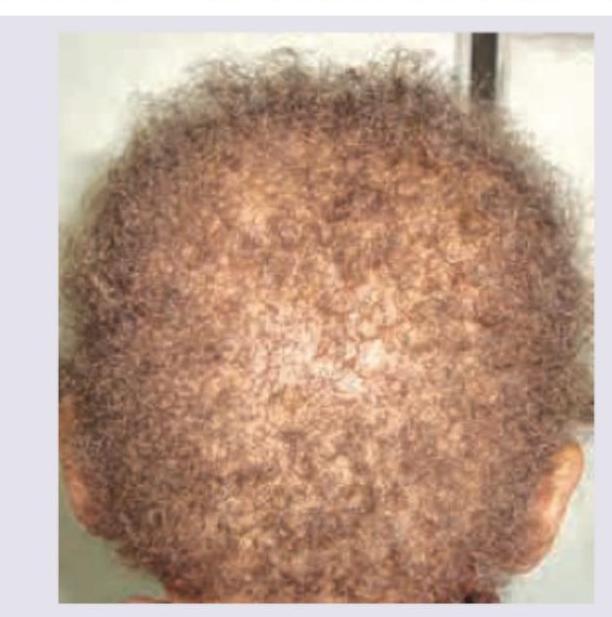
The patient shown below has curly easily breakable hair. Which is correct about the condition?
Biochemical screening of newborn infants by heel-prick blood samples is performed by using the
Which one of the following conditions is NOT inborn error of metabolism?
Practice by Chapter
Single Gene Disorders
Practice Questions
Biochemical Diagnosis of Genetic Disorders
Practice Questions
Inborn Errors of Metabolism
Practice Questions
Lysosomal Storage Diseases
Practice Questions
Glycogen Storage Diseases
Practice Questions
Disorders of Lipoprotein Metabolism
Practice Questions
Disorders of Purine and Pyrimidine Metabolism
Practice Questions
Hemoglobinopathies
Practice Questions
Porphyrias
Practice Questions
Biochemical Markers for Disease Diagnosis
Practice Questions
Newborn Screening for Genetic Disorders
Practice Questions
Enzyme Replacement Therapy
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app