
Genetic Disorders and Biochemical Pathology — MCQs

On this page
What is the most common cause of congenital adrenal hyperplasia?
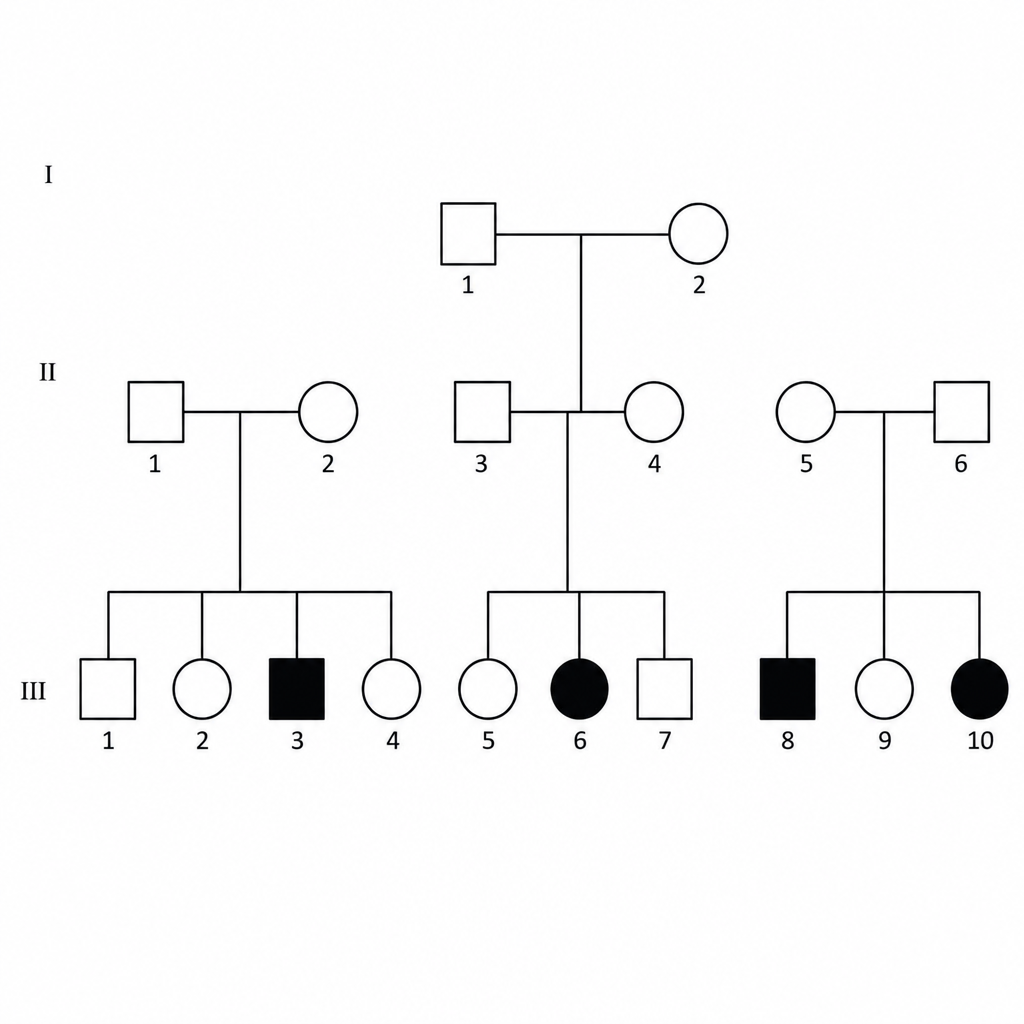
The following pedigree is associated with which of the following conditions?
NARP syndrome is a part of which of the following group of disorders?
A 54-year-old male with acute lymphocytic leukemia develops a blast crisis and is treated with intensive systemic chemotherapy. Following treatment, the patient will be at increased risk for the development of which of the following?
Which of the following is NOT a mitochondrial disorder?
What is the biochemical defect in Zellweger syndrome?
All of the following statements about the inheritance of Myotonic Dystrophy are true, except?
A young adult presents with progressive intellectual deterioration, weakness, ataxia, and seizures. Laboratory tests demonstrate an abnormality of an important mitochondrial enzyme. Which of the following conditions is this person most likely suffering from?
Which of the following statements is FALSE regarding G6PD deficiency?
A male infant's parents report that male children over three generations in the mother's family have been affected by a progressive disorder involving multiple organ systems. These children had coarse facial features, corneal clouding, joint stiffness, hepatosplenomegaly, and mental retardation, and many died in childhood. At autopsy, some of the children had subendothelial coronary arterial deposits that caused myocardial infarction. Laboratory testing of the infant shows increased urinary excretion of mucopolysaccharides. Bone marrow biopsy reveals that the accumulated mucopolysaccharides are found in macrophages ('balloon cells' filled with minute vacuoles). Which of the following enzyme deficiencies is most likely to be seen in this infant?
Practice by Chapter
Single Gene Disorders
Practice Questions
Biochemical Diagnosis of Genetic Disorders
Practice Questions
Inborn Errors of Metabolism
Practice Questions
Lysosomal Storage Diseases
Practice Questions
Glycogen Storage Diseases
Practice Questions
Disorders of Lipoprotein Metabolism
Practice Questions
Disorders of Purine and Pyrimidine Metabolism
Practice Questions
Hemoglobinopathies
Practice Questions
Porphyrias
Practice Questions
Biochemical Markers for Disease Diagnosis
Practice Questions
Newborn Screening for Genetic Disorders
Practice Questions
Enzyme Replacement Therapy
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app