
Genetic Disorders and Biochemical Pathology — MCQs

On this page
What is true about Gilbert syndrome?
Bilirubin UDP glucuronyl transferase activity is absent in which of the following conditions?
Very low activity of adenosine deaminase in red blood cells and high levels of dATP is consistent with which diagnosis?
A screening test for phenylketonuria (PKU) is performed on umbilical cord blood from a fair-skinned blond, blue-eyed infant born to dark-complexioned parents. The test is reported as negative, and no dietary restrictions are imposed. At 1 year of age, the child is seen again, this time with obvious signs of severe mental retardation, and a diagnosis of PKU is made. The diagnosis was missed at birth because:
Congenital adrenal hyperplasia is most likely a result of which of the following?
A single gene defect causing multiple unrelated problems is termed as the following?
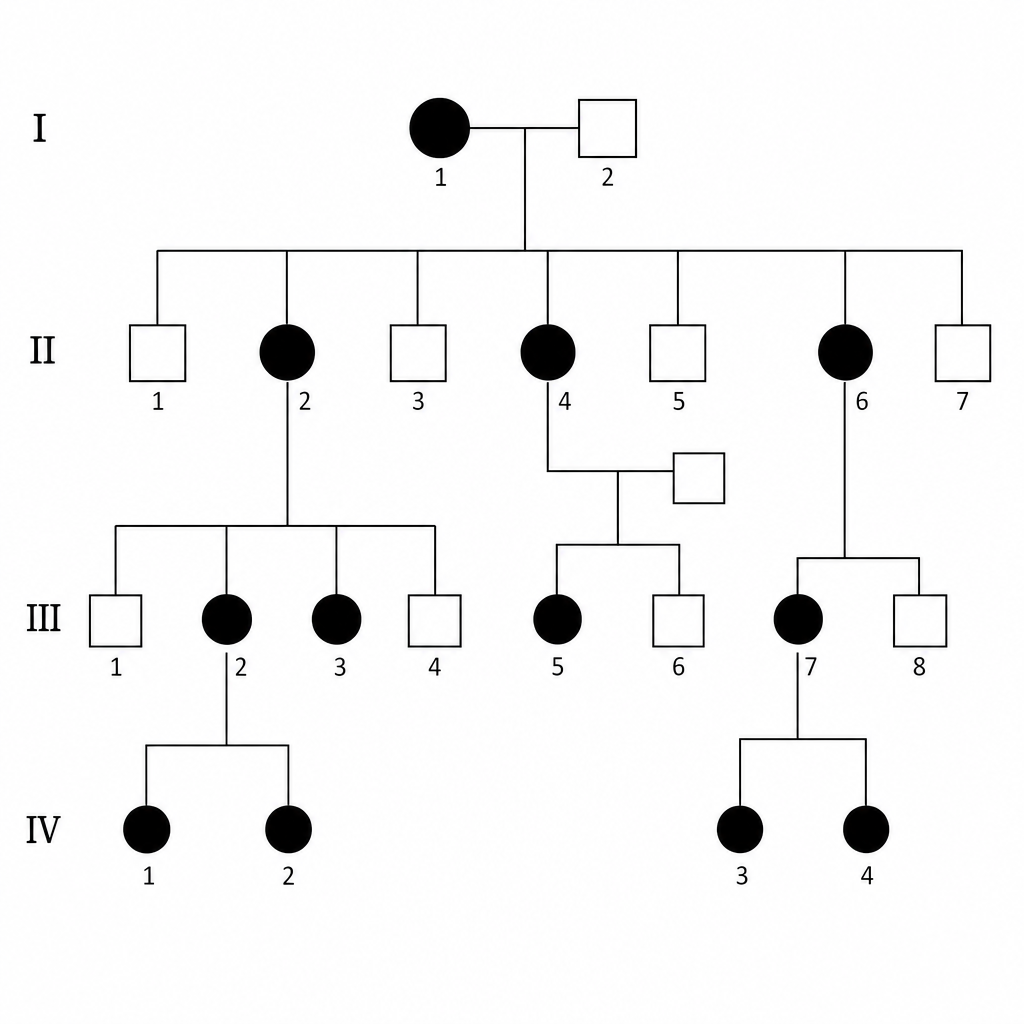
Find the type of inheritance?
Which of the following is FALSE regarding hereditary fructose intolerance?
Cirrhosis can be seen in all of the following metabolic diseases EXCEPT?
Which of the following statements about mitochondrial disorders is FALSE?
Practice by Chapter
Single Gene Disorders
Practice Questions
Biochemical Diagnosis of Genetic Disorders
Practice Questions
Inborn Errors of Metabolism
Practice Questions
Lysosomal Storage Diseases
Practice Questions
Glycogen Storage Diseases
Practice Questions
Disorders of Lipoprotein Metabolism
Practice Questions
Disorders of Purine and Pyrimidine Metabolism
Practice Questions
Hemoglobinopathies
Practice Questions
Porphyrias
Practice Questions
Biochemical Markers for Disease Diagnosis
Practice Questions
Newborn Screening for Genetic Disorders
Practice Questions
Enzyme Replacement Therapy
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app