
Genetic Disorders and Biochemical Pathology — MCQs

On this page
A patient presented to the OPD with liver damage. The picture depicts the patient having their eyes examined. Which of the following substances is responsible for this condition?
A child has elevated liver enzyme levels. A ring-like structure is noted on ocular examination. Which of the following is the cause for this?
A family pedigree chart is given below. Identify the mode of inheritance of this condition.
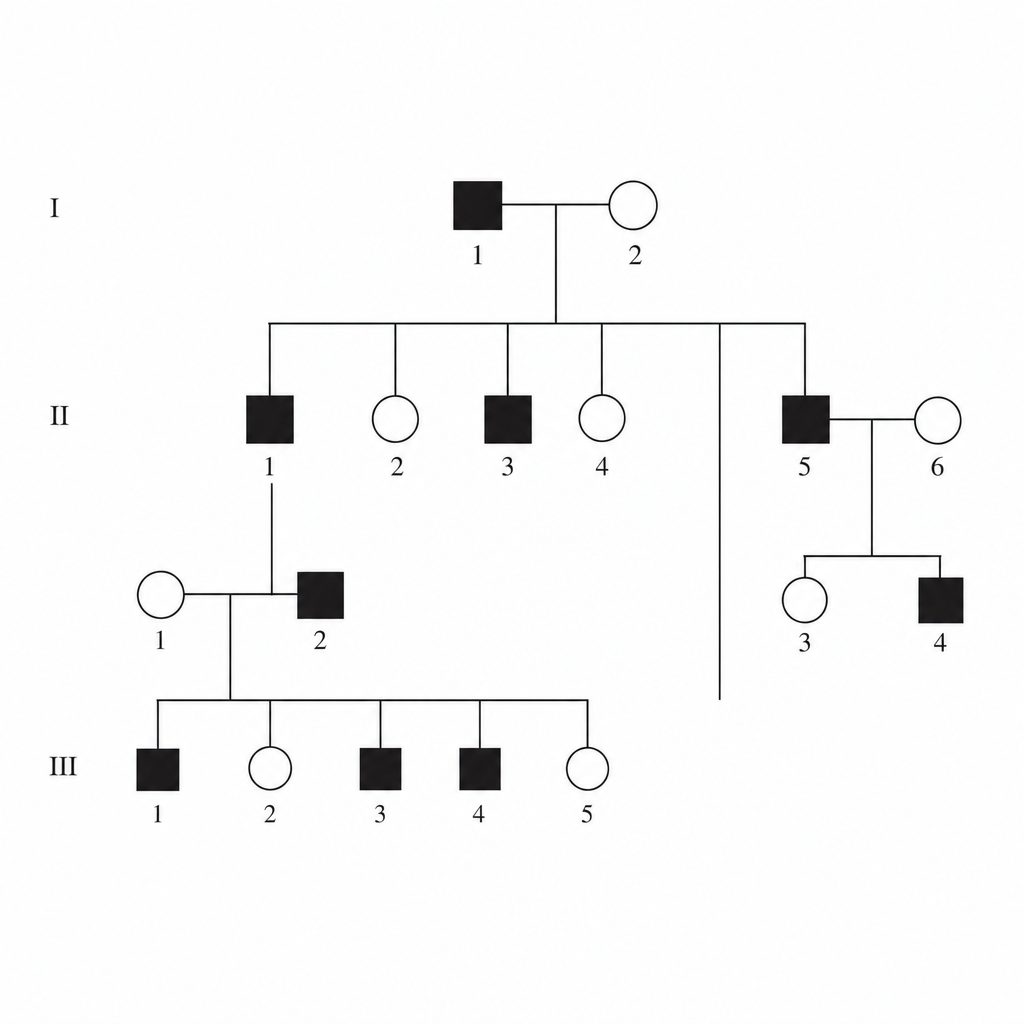
A family pedigree chart is given below. Identify the mode of inheritance of this condition. 
A deficiency of Glucose-6-Phosphatase is associated with which of the following bilirubin patterns?
The pedigree diagram of a family is shown below. Affected individuals present with progressive external ophthalmoplegia, pigmentary retinopathy, and cardiac conduction defects. Based on the pedigree and clinical features, what is the most likely diagnosis?
A 6-month-old infant presents with recurrent infections and failure to thrive. Laboratory investigations reveal a deficiency of adenosine deaminase (ADA). Which of the following immunodeficiency disorders is most likely associated?
A child presents with developmental delay and coarse facial features. Enzyme assay reveals a deficiency of α-L-iduronidase. Which of the following substances is most likely to accumulate in this condition?
Which of the following is a mitochondrial inheritance disorder?
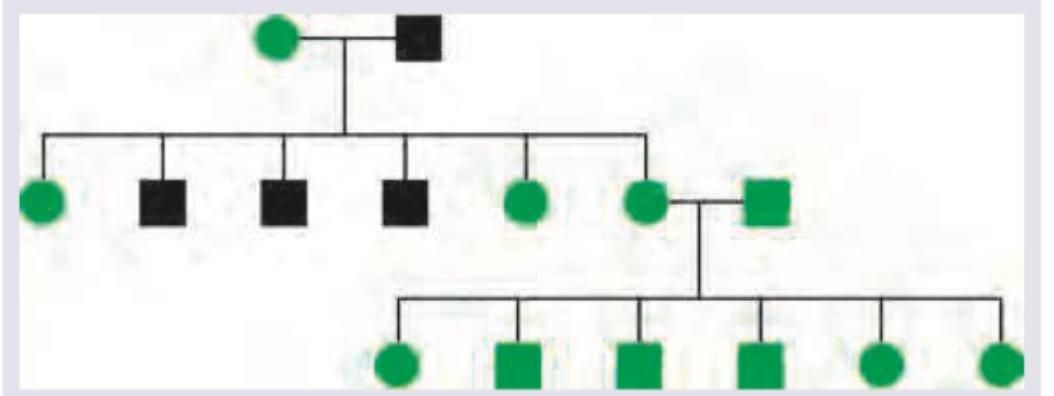
Identify the pattern of inheritance shown below.
Practice by Chapter
Single Gene Disorders
Practice Questions
Biochemical Diagnosis of Genetic Disorders
Practice Questions
Inborn Errors of Metabolism
Practice Questions
Lysosomal Storage Diseases
Practice Questions
Glycogen Storage Diseases
Practice Questions
Disorders of Lipoprotein Metabolism
Practice Questions
Disorders of Purine and Pyrimidine Metabolism
Practice Questions
Hemoglobinopathies
Practice Questions
Porphyrias
Practice Questions
Biochemical Markers for Disease Diagnosis
Practice Questions
Newborn Screening for Genetic Disorders
Practice Questions
Enzyme Replacement Therapy
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app