
Genetic Disorders and Biochemical Pathology — MCQs

On this page
All are important pathological features noted in ATP7B gene mutation, EXCEPT?
Enzyme replacement therapy is available for the treatment of which of the following disorders?
Which enzyme deficiency is seen in genetic diseases like Tay-Sachs disease?
A 48-year-old lady presented with hepatosplenomegaly and pancytopenia. On microscopic examination of bone marrow cells, a crumpled tissue paper appearance is seen. Which product is likely to have accumulated?
Which one of the following inherited conditions causes direct hyperbilirubinemia?
The syndrome of apparent mineralocorticoid excess is due to deficiency of which enzyme?
A 6-month-old baby presents with recurrent seizures, developmental delay, alopecia, and scaly skin rashes. Investigations reveal metabolic acidosis, elevated lactates, and ketonuria. What is the most likely underlying enzyme deficiency?
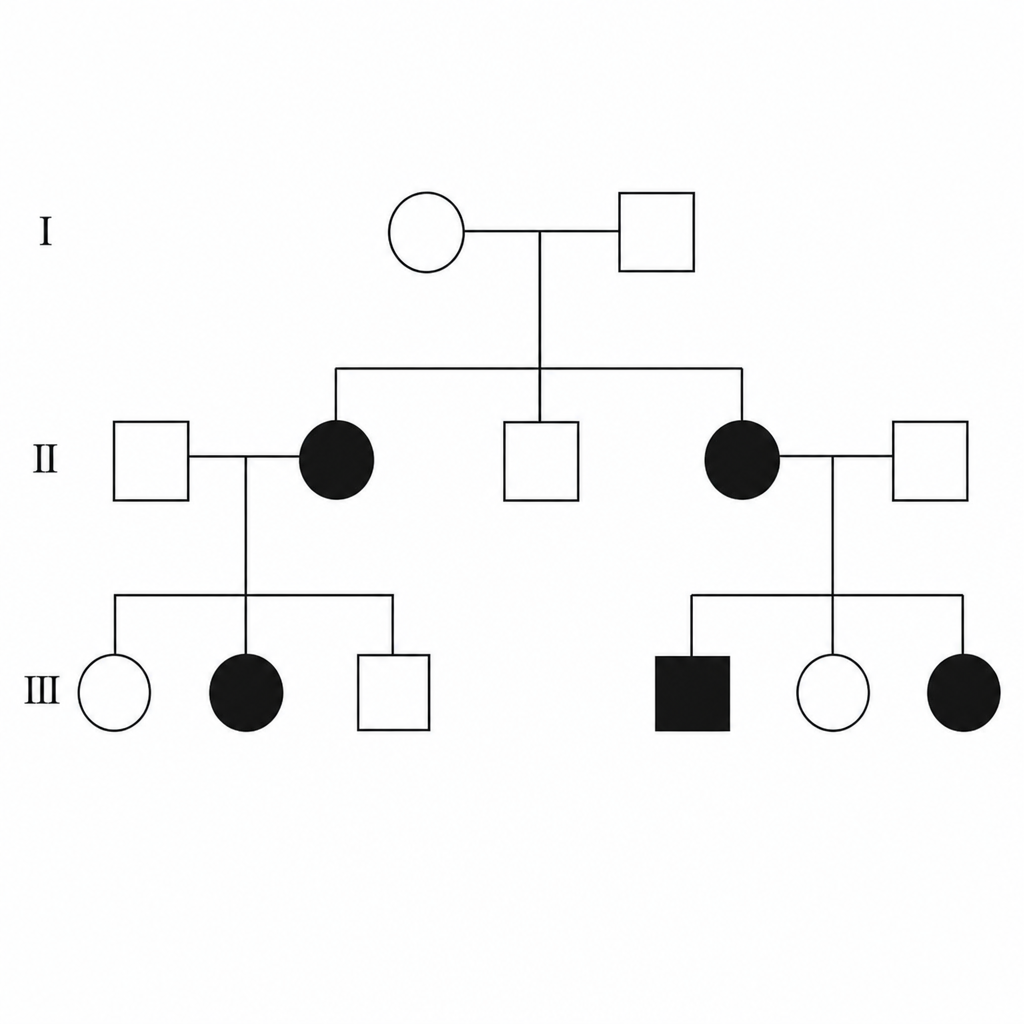
Analyze the following pedigree and determine the mode of inheritance.
Hypoceruloplasminemia is associated with which abnormality?
Tomcat urine odor is characteristic of which of the following conditions?
Practice by Chapter
Single Gene Disorders
Practice Questions
Biochemical Diagnosis of Genetic Disorders
Practice Questions
Inborn Errors of Metabolism
Practice Questions
Lysosomal Storage Diseases
Practice Questions
Glycogen Storage Diseases
Practice Questions
Disorders of Lipoprotein Metabolism
Practice Questions
Disorders of Purine and Pyrimidine Metabolism
Practice Questions
Hemoglobinopathies
Practice Questions
Porphyrias
Practice Questions
Biochemical Markers for Disease Diagnosis
Practice Questions
Newborn Screening for Genetic Disorders
Practice Questions
Enzyme Replacement Therapy
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app