
Genetic Disorders and Biochemical Pathology — MCQs

On this page
A 10-year-old child presents with symptoms suggestive of pellagra, including chronic diarrhea, a red scaly rash, and mild cerebellar ataxia. The child's diet is adequate in protein and niacin. A sister has a similar presentation. Chemical analysis of the patient's urine shows large amounts of free amino acids. What is the most likely diagnosis?
Which one of the following is not a mitochondrial disorder?
All are true about Fabry disease, EXCEPT:
Which treatment is contraindicated in hypophosphatasia?
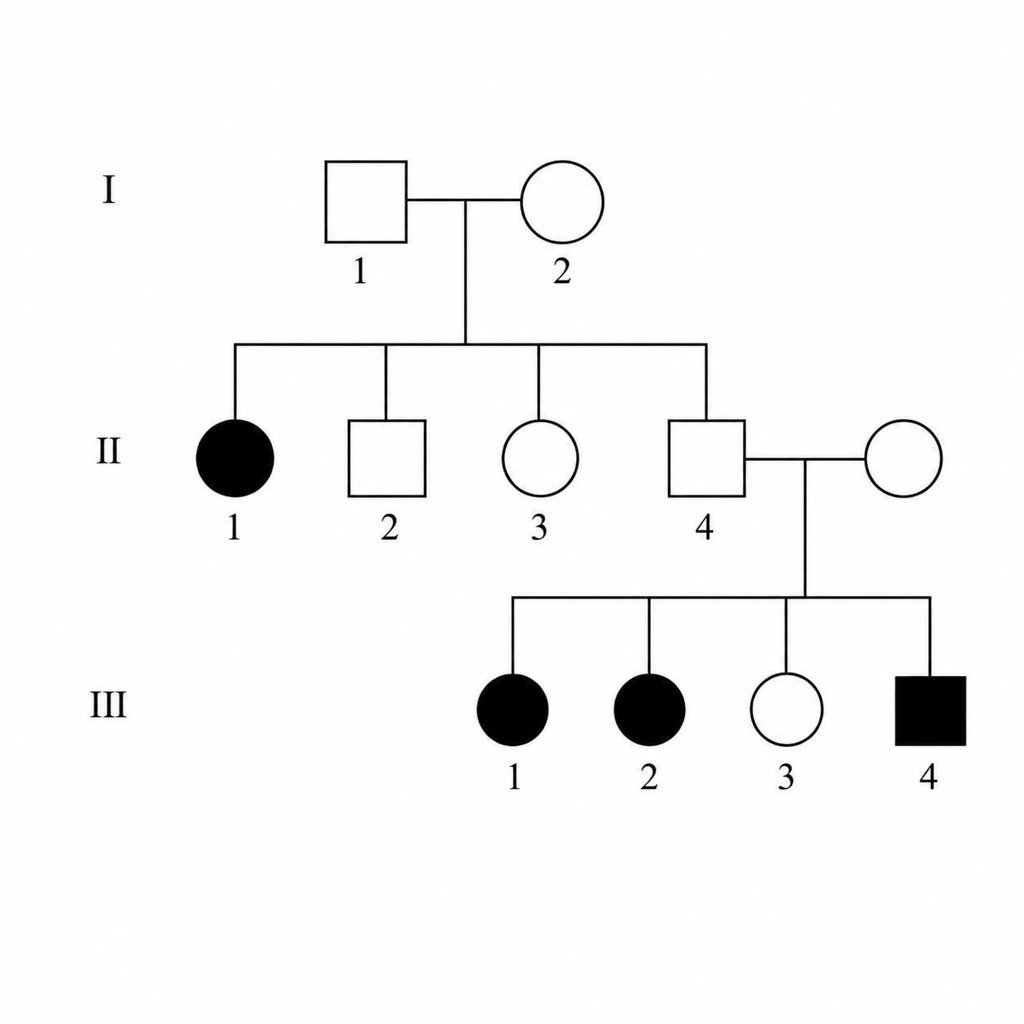
Which of the following is the most likely inheritance pattern in the given pedigree?
In Crigler-Najjar syndrome type II, what is the primary defect?
Which one of the following is not a feature of Phenylketonuria?
All of the following are X-linked recessive disorders except?
Pyrimidine 5'-Nucleotidase deficiency presents clinically as:
The genetic defect in Dubin-Johnson Syndrome is a mutation in which of the following?
Practice by Chapter
Single Gene Disorders
Practice Questions
Biochemical Diagnosis of Genetic Disorders
Practice Questions
Inborn Errors of Metabolism
Practice Questions
Lysosomal Storage Diseases
Practice Questions
Glycogen Storage Diseases
Practice Questions
Disorders of Lipoprotein Metabolism
Practice Questions
Disorders of Purine and Pyrimidine Metabolism
Practice Questions
Hemoglobinopathies
Practice Questions
Porphyrias
Practice Questions
Biochemical Markers for Disease Diagnosis
Practice Questions
Newborn Screening for Genetic Disorders
Practice Questions
Enzyme Replacement Therapy
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app