
Genetic Disorders and Biochemical Pathology — MCQs

On this page
Which enzyme deficiency is most commonly responsible for the presence of a long clitoris and fused vagina?
All of the following are associated with non-ketotic hypoglycemia, EXCEPT?
Alpha-1-antitrypsin deficiency is associated with a gene located on which chromosome?
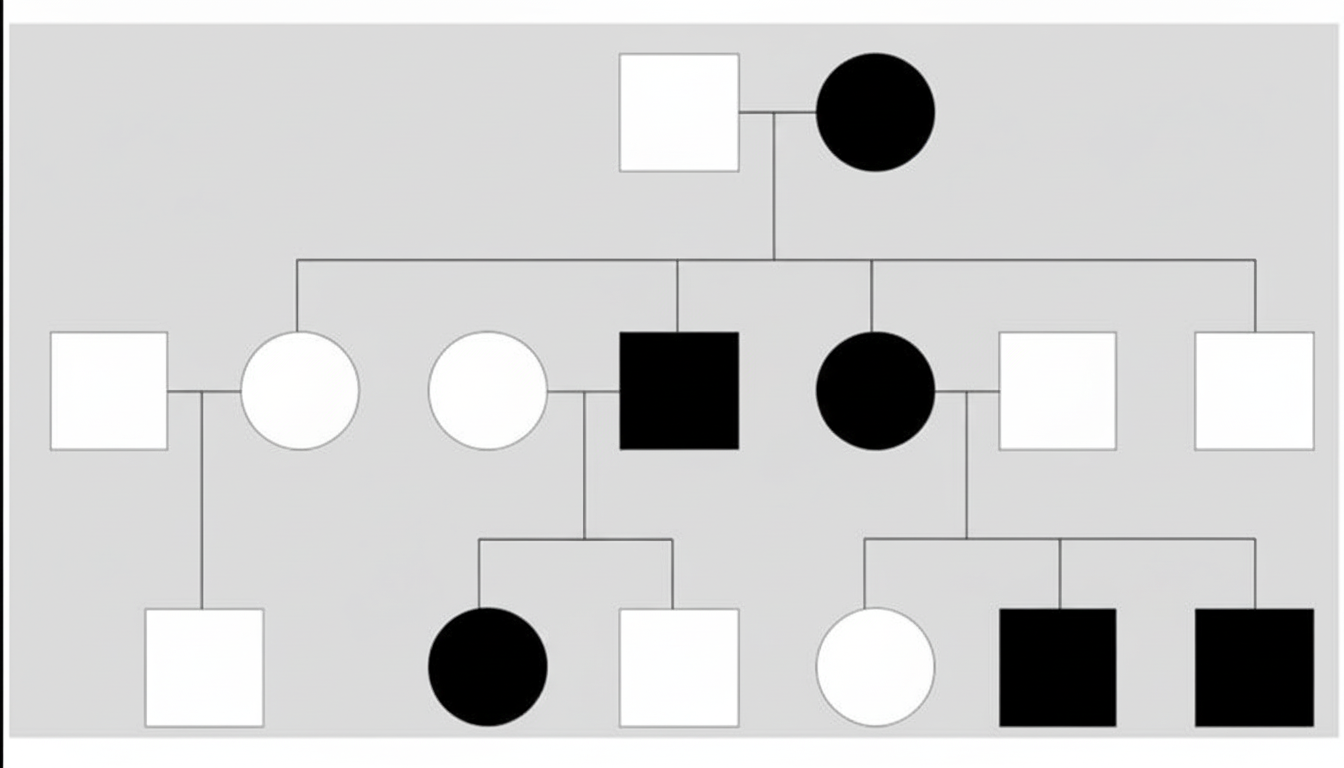
The following pedigree represents which of the following genetic disorders?
Which of the following diseases is NOT a mitochondrial disorder?
Friedreich's ataxia is caused by which of the following genetic mechanisms?
All of the following conditions have autosomal dominant inheritance except?
What is true about Crigler-Najjar type II syndrome?
Which of the following is not included under Garrod's tetrad?
Nitisinone is an example of which of the following therapeutic approaches?
Practice by Chapter
Single Gene Disorders
Practice Questions
Biochemical Diagnosis of Genetic Disorders
Practice Questions
Inborn Errors of Metabolism
Practice Questions
Lysosomal Storage Diseases
Practice Questions
Glycogen Storage Diseases
Practice Questions
Disorders of Lipoprotein Metabolism
Practice Questions
Disorders of Purine and Pyrimidine Metabolism
Practice Questions
Hemoglobinopathies
Practice Questions
Porphyrias
Practice Questions
Biochemical Markers for Disease Diagnosis
Practice Questions
Newborn Screening for Genetic Disorders
Practice Questions
Enzyme Replacement Therapy
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app