
Genetic Disorders and Biochemical Pathology — MCQs

On this page
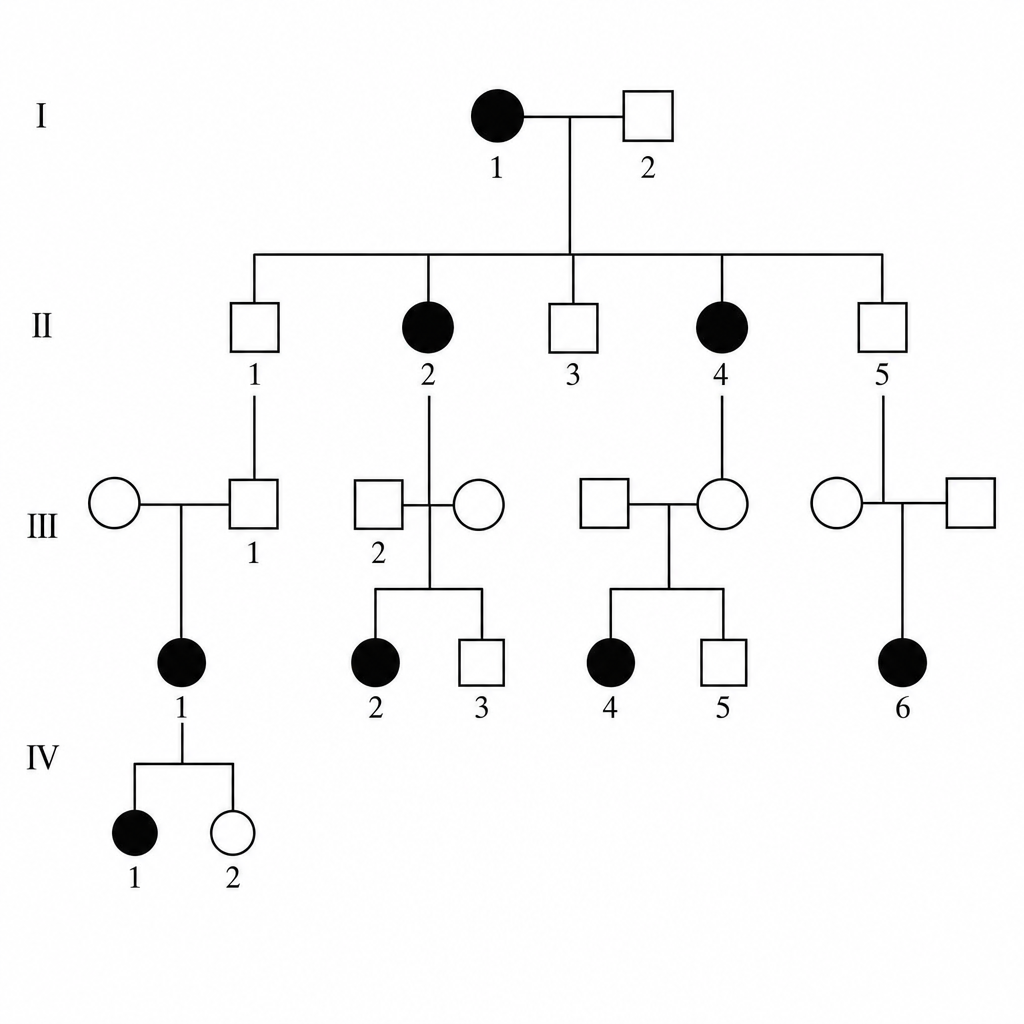
Which of the following is the most likely inheritance pattern shown in the pedigree?
Which chromosome is associated with Alzheimer's disease?
Which genetic disorder is caused by a microdeletion on chromosome 15?
Complete deficiency of UDP glucuronyl transferase (UGT) is seen in which of the following conditions?
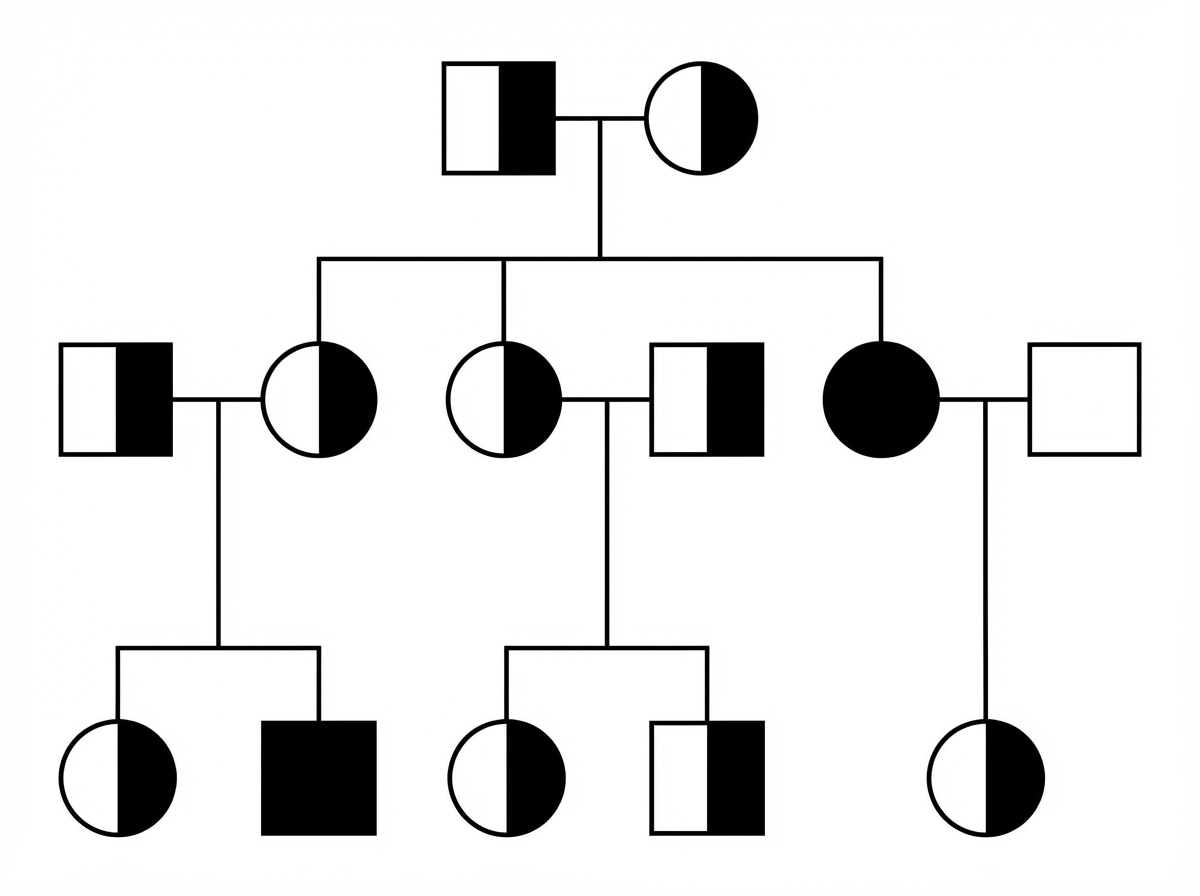
Examine the given pedigree chart. Which one of the following diseases is the most likely for this situation?
AST/ALT ratio > 2 is seen in deficiency of which enzyme?
'I' cells disease is due to a defect in which cellular organelle?
The gene involved in Rett's syndrome is:
Which of the following genetic factors does NOT increase susceptibility and modify the severity of pancreatic injury in acute pancreatitis?
Which hormone levels are increased in Prader-Willi syndrome?
Practice by Chapter
Single Gene Disorders
Practice Questions
Biochemical Diagnosis of Genetic Disorders
Practice Questions
Inborn Errors of Metabolism
Practice Questions
Lysosomal Storage Diseases
Practice Questions
Glycogen Storage Diseases
Practice Questions
Disorders of Lipoprotein Metabolism
Practice Questions
Disorders of Purine and Pyrimidine Metabolism
Practice Questions
Hemoglobinopathies
Practice Questions
Porphyrias
Practice Questions
Biochemical Markers for Disease Diagnosis
Practice Questions
Newborn Screening for Genetic Disorders
Practice Questions
Enzyme Replacement Therapy
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app