
Genetic Disorders and Biochemical Pathology — MCQs

On this page
An infant is unable to feed properly, is weak, and not gaining weight. The mother reports multiple episodes of urination with crying each time the baby passes urine. She also notes that the baby often smells of rotten fish in his urine and sweat. Which of the following would you test for in the infant's urine?
A 9-month-old infant presented with recurrent infections. Investigations revealed a near absence of B and T cells and a significantly diminished thymic shadow on chest X-ray. Which of the following metabolites would be elevated in this patient?
For which of the following diseases is enzyme replacement therapy available?
Barth syndrome is due to a defect in which of the following?
Different mutations in the same genetic locus causing similar or identical phenotypes is termed as?
All of the following are examples of uniparental disomy except?
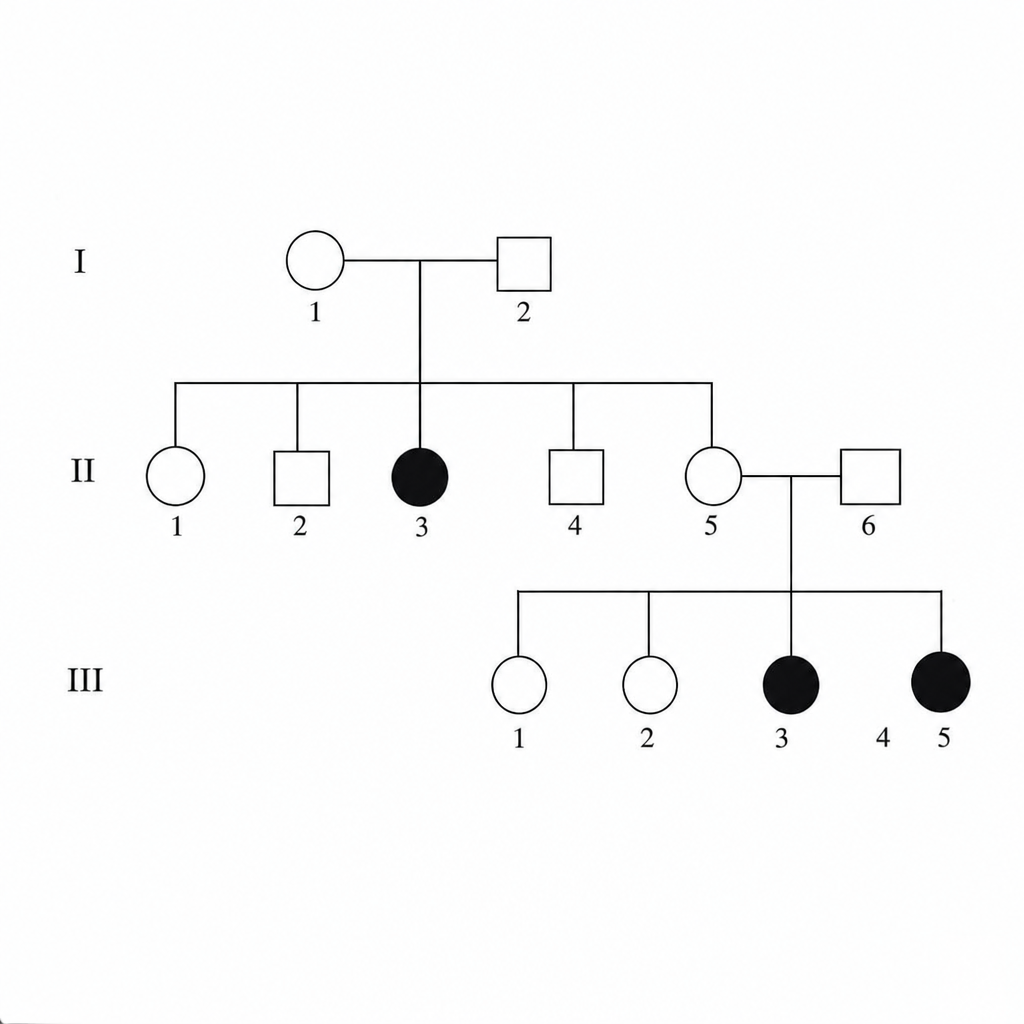
The provided diagram indicates what type of inheritance?
Menke's disease is due to a defect in the metabolism of which element?
Which of the following is not characteristic of vitamin-D resistant rickets?
Unconjugated hyperbilirubinemia is seen in which of the following conditions?
Practice by Chapter
Single Gene Disorders
Practice Questions
Biochemical Diagnosis of Genetic Disorders
Practice Questions
Inborn Errors of Metabolism
Practice Questions
Lysosomal Storage Diseases
Practice Questions
Glycogen Storage Diseases
Practice Questions
Disorders of Lipoprotein Metabolism
Practice Questions
Disorders of Purine and Pyrimidine Metabolism
Practice Questions
Hemoglobinopathies
Practice Questions
Porphyrias
Practice Questions
Biochemical Markers for Disease Diagnosis
Practice Questions
Newborn Screening for Genetic Disorders
Practice Questions
Enzyme Replacement Therapy
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app