
Genetic Disorders and Biochemical Pathology — MCQs

On this page
Gaucher disease is inherited in which pattern?
Which of the following genetic disorders exclusively affects males?
An affected male infant born to unaffected parents could be an example of all of the following inheritance patterns, except?
Which of the following is NOT a lysosomal storage disorder?
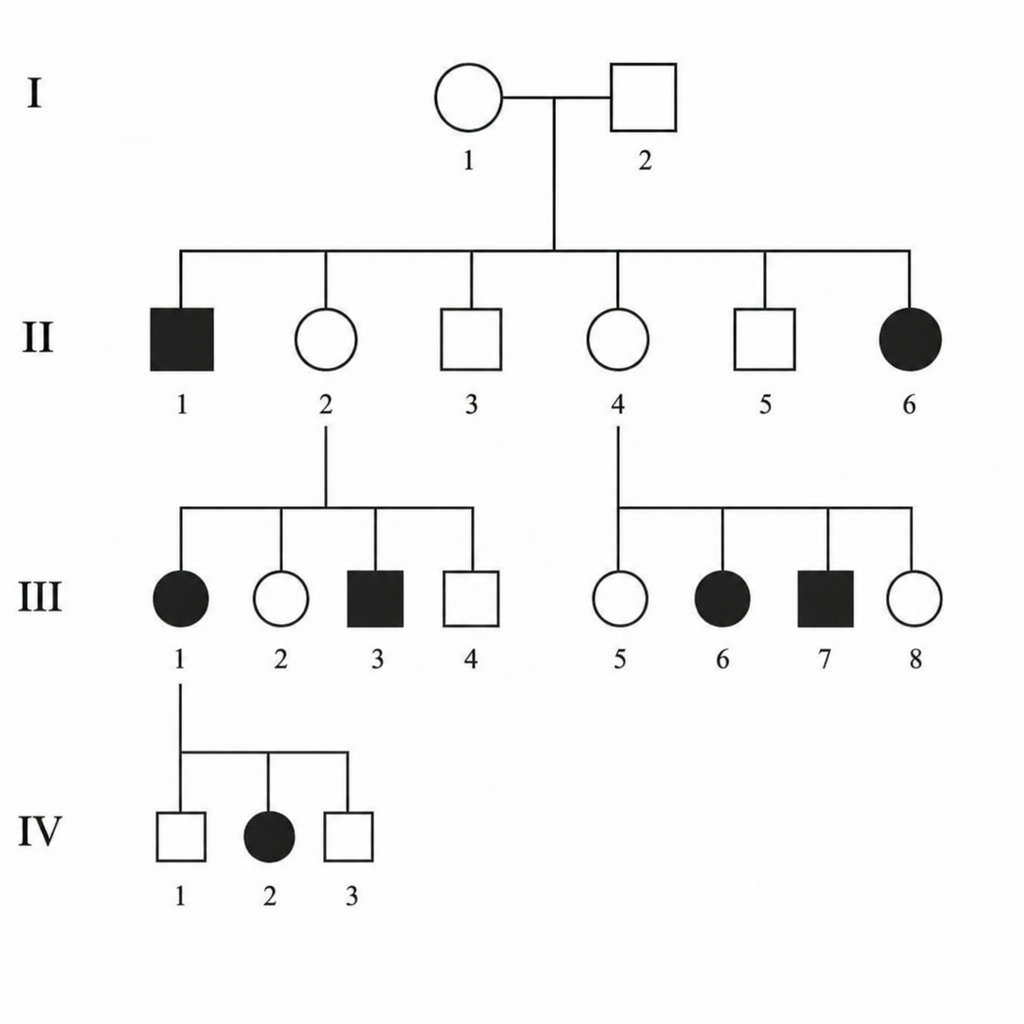
Which type of inheritance is indicated?
Defect in which of the following proteins leads to Rett syndrome?
Which of the following is NOT a recognized association?
A woman who suffers from frequent and severe migraine headaches has had five children, all of whom have experienced, beginning between the ages of 8 and 12, stroke-like episodes compounded with exercise intolerance and lactic acidosis. The father of the children does not suffer from migraines, nor is he exercise intolerant. A target of a mutation that can explain these findings is most likely which one of the following?
The gene for Wilson's disease is located on which chromosome?
Which chromosome is associated with cystic fibrosis?
Practice by Chapter
Single Gene Disorders
Practice Questions
Biochemical Diagnosis of Genetic Disorders
Practice Questions
Inborn Errors of Metabolism
Practice Questions
Lysosomal Storage Diseases
Practice Questions
Glycogen Storage Diseases
Practice Questions
Disorders of Lipoprotein Metabolism
Practice Questions
Disorders of Purine and Pyrimidine Metabolism
Practice Questions
Hemoglobinopathies
Practice Questions
Porphyrias
Practice Questions
Biochemical Markers for Disease Diagnosis
Practice Questions
Newborn Screening for Genetic Disorders
Practice Questions
Enzyme Replacement Therapy
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app