NEET-PG 2024 — Biochemistry
20 Previous Year Questions with Answers & Explanations
A 45-year-old patient presents with symptoms of anemia, depigmented hair, and myelopathy. Which of the following mineral deficiencies is most likely associated with this clinical presentation?
An infant presents with vomiting after feeding. Benedict's test was positive for a non-glucose reducing substance. What is the most likely diagnosis?
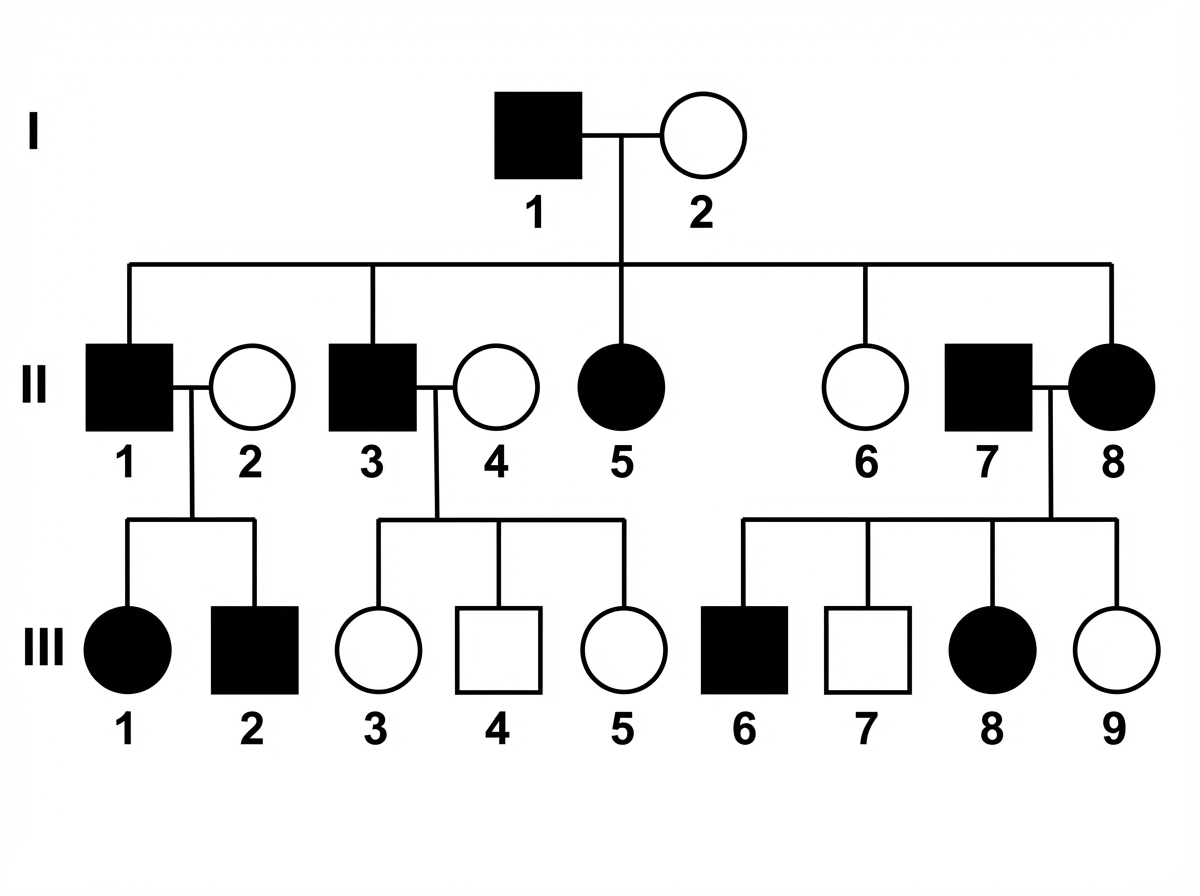
Which disease will show the mode of inheritance depicted in this pedigree?
A mother brings her 14 -year-old mentally retarded son to the OPD, who is suffering from joint pain and has an irresistible urge to bite his fingers and lips. His serum uric acid level was found to be elevated. What is the disorder?
A patient presents with symptoms of dermatitis, dementia, and cognitive decline. Which micronutrient deficiency is most likely responsible?
A 5-year-old girl was washing her doll with shampoo containing rotenone. Her mother noticed her in an unconscious state. Which enzyme is inhibited by the above chemical?
A patient with high triglycerides (TG) esterified with long-chain fatty acids (LCFA) presents with fatigue, and a biopsy of the muscle shows fat vacuoles. What is the most likely diagnosis?
A patient presents with xanthomas on the Achilles tendon. What is the most likely diagnosis?
What is the source of Vitamin B1 (Thiamine)?

A patient has multiple tendon xanthomas. Serum cholesterol ( $398 \mathrm{mg} / \mathrm{dL}$ ) and LDL ( 220 $\mathrm{mg} / \mathrm{dL}$ ) were found to be elevated. What is the most likely defect?
NEET-PG 2024 - Biochemistry NEET-PG Practice Questions and MCQs
Question 1: A 45-year-old patient presents with symptoms of anemia, depigmented hair, and myelopathy. Which of the following mineral deficiencies is most likely associated with this clinical presentation?
- A. Copper (Correct Answer)
- B. Iron
- C. Fluoride
- D. Zinc
- E. Selenium
Explanation: ***Copper*** - **Copper deficiency** can lead to anemia due to its role in iron metabolism, **depigmented hair** (achromotrichia) due to impaired melanin synthesis, and **myelopathy** due to its involvement in maintaining the myelin sheath. - Symptoms often mimic those of **vitamin B12 deficiency**, including neurological manifestations like ataxia and spasticity. *Iron* - **Iron deficiency** is the most common cause of anemia but does not typically cause **depigmented hair** or **myelopathy**. - Its neurological symptoms are usually limited to **restless legs syndrome** and **pica**, not demyelination. *Fluoride* - **Fluoride deficiency** is primarily associated with an increased risk of dental caries and does not cause anemia, hair depigmentation, or myelopathy. - Excessive intake (fluorosis) can lead to **bone and tooth abnormalities**. *Zinc* - **Zinc deficiency** can cause immune dysfunction, **dermatitis**, impaired wound healing, and growth retardation. - It may rarely cause anemia in severe cases but does not typically cause hair depigmentation or myelopathy as primary symptoms. *Selenium* - **Selenium deficiency** is associated with **Keshan disease** (cardiomyopathy) and **Kashin-Beck disease** (osteoarthropathy). - While it can cause muscle weakness and fatigue, it does not typically present with the specific triad of anemia, hair depigmentation, and myelopathy seen in copper deficiency.
Question 2: An infant presents with vomiting after feeding. Benedict's test was positive for a non-glucose reducing substance. What is the most likely diagnosis?
- A. Galactosemia due to GAL-1-P Uridyl Transferase enzyme deficiency (Correct Answer)
- B. Fructosuria due to Fructokinase deficiency
- C. Hereditary fructose intolerance due to Aldolase B deficiency
- D. Primary lactose intolerance
- E. Glycogen storage disease due to Glucose-6-phosphatase deficiency
Explanation: ***Galactosemia due to GAL-1-P Uridyl Transferase enzyme deficiency*** - Vomiting after feeding in an infant, coupled with a **positive Benedict's test** for a **non-glucose reducing substance**, is highly indicative of galactosemia. The accumulation of **galactose-1-phosphate** and **galactitol** leads to toxicity and symptoms. - This enzyme deficiency, causing **classic galactosemia**, prevents the proper metabolism of **galactose**, leading to its buildup. *Fructosuria due to Fructokinase deficiency* - This condition is a **benign metabolic disorder** with no significant clinical symptoms. - While it would lead to fructose in the urine, the infant would not typically present with **vomiting after feeding**. *Hereditary fructose intolerance due to Aldolase B deficiency* - Symptoms usually appear after the introduction of **fructose-containing foods** into the diet, causing severe hypoglycemia and vomiting. - The positive Benedict's test in this scenario would typically indicate a reducing substance in the urine, while fructose intolerance is characterized by **hypoglycemia** and metabolic crises upon fructose ingestion. *Glycogen storage disease due to Glucose-6-phosphatase deficiency* - This disorder primarily causes **hypoglycemia** and liver enlargement, not primarily vomiting after feeding due to a **non-glucose reducing substance**. - Glucose-6-phosphatase deficiency (Von Gierke's disease) leads to an inability to release **glucose from glycogen** and causes severe hypoglycemia, often requiring frequent feeding. *Primary lactose intolerance* - While lactose intolerance can cause vomiting and gastrointestinal symptoms, it is **extremely rare in infants** (primary lactose intolerance is a late-onset condition). - Lactose would be a reducing sugar, but the key differentiator is that **galactose** (from galactosemia) is the non-glucose reducing substance detected in this case, along with the typical **toxic presentation** in neonates.
Question 3: Which disease will show the mode of inheritance depicted in this pedigree?
- A. Achondroplasia (Correct Answer)
- B. Prader-Willi syndrome
- C. Wilson disease
- D. Wiskott-Aldrich syndrome
- E. Hemophilia A
Explanation: ***Achondroplasia*** - This pedigree shows an **autosomal dominant** inheritance pattern, characterized by affected individuals in every generation, affected offspring with at least one affected parent, and direct transmission from father to son (which **rules out X-linked**). Achondroplasia is an autosomal dominant disorder. - The presence of **affected individuals (shaded squares/circles)** in each successive generation and the 1:1 ratio of affected to unaffected offspring from affected parents mating with unaffected individuals supports autosomal dominant inheritance. *Prader-Willi syndrome* - This syndrome is caused by **genomic imprinting** or a chromosomal deletion on chromosome 15, typically inherited from the father. It does not follow a simple Mendelian dominant or recessive inheritance pattern. - While it has a genetic basis, its inheritance pattern is **complex** and involves specific parental origin of the genetic defect, unlike the clear autosomal dominant pattern shown. *Wilson disease* - Wilson disease is an **autosomal recessive** disorder, meaning affected individuals inherit two copies of the mutated gene (one from each parent). - This pedigree does not show skipped generations or unaffected parents having affected offspring, which would be characteristic of **autosomal recessive** inheritance. *Wiskott-Aldrich syndrome* - Wiskott-Aldrich syndrome is an **X-linked recessive** disorder. This means it primarily affects males, and affected fathers cannot pass the trait to their sons. - The pedigree shows **affected females** (shaded circles) and **father-to-son transmission** (e.g., father in second generation to son in third generation, assuming the leftmost branch is the paternal line), which rules out X-linked inheritance. *Hemophilia A* - Hemophilia A is an **X-linked recessive** disorder affecting the Factor VIII gene, predominantly affecting males. - Similar to Wiskott-Aldrich syndrome, the presence of **father-to-son transmission** in the pedigree rules out X-linked inheritance patterns, as affected fathers cannot pass X-linked traits to their sons.
Question 4: A mother brings her 14 -year-old mentally retarded son to the OPD, who is suffering from joint pain and has an irresistible urge to bite his fingers and lips. His serum uric acid level was found to be elevated. What is the disorder?
- A. Lesch-Nyhan syndrome (Correct Answer)
- B. Xanthine oxidase deficiency
- C. CPS II defect
- D. Thymidylate synthetase
- E. Von Gierke disease
Explanation: ***Lesch-Nyhan syndrome*** - This syndrome is characterized by **X-linked recessive inheritance** and a deficiency of the enzyme **hypoxanthine-guanine phosphoribosyltransferase (HGPRT)**, leading to an overproduction of uric acid. - Clinical manifestations include **hyperuricemia**, **gout-like arthritis**, **neurological dysfunction** (mental retardation, dystonia), and a distinctive feature of **self-mutilating behaviors** such as biting fingers and lips. *Xanthine oxidase deficiency* - This condition leads to an accumulation of **hypoxanthine** and **xanthine** due to impaired conversion to uric acid. - While it can cause kidney stones and some joint pain, it typically results in *low* serum uric acid levels and does not present with the characteristic self-mutilating behavior or severe neurological deficits seen in Lesch-Nyhan. *CPS II defect* - **Carbamoyl phosphate synthetase II (CPS II)** is involved in *pyrimidine synthesis*, not purine metabolism. - A defect in CPS II would lead to issues with pyrimidine production and typically presents with megaloblastic anemia or other symptoms related to pyrimidine deficiency, not hyperuricemia or self-mutilation. *Thymidylate synthetase* - **Thymidylate synthetase (TS)** is an enzyme critical for the synthesis of **thymidylate**, a precursor for DNA synthesis. - Inhibition or deficiency of TS is primarily associated with DNA replication issues, megaloblastic anemia, and is a target for chemotherapy, not with hyperuricemia or the neurological and behavioral symptoms described. *Von Gierke disease* - **Von Gierke disease** (Glycogen storage disease type Ia) is caused by deficiency of **glucose-6-phosphatase**, leading to impaired conversion of glucose-6-phosphate to glucose. - While it can cause **hyperuricemia** due to increased uric acid production from accelerated purine degradation, the clinical presentation is distinct with **hepatomegaly**, **hypoglycemia**, **lactic acidosis**, and growth retardation. - It does **not** present with self-mutilating behavior or the severe neurological dysfunction characteristic of Lesch-Nyhan syndrome.
Question 5: A patient presents with symptoms of dermatitis, dementia, and cognitive decline. Which micronutrient deficiency is most likely responsible?
- A. Niacin (Correct Answer)
- B. Thiamine
- C. Tryptophan
- D. Retinol
- E. Riboflavin
Explanation: ***Niacin*** - The classic triad of symptoms known as the "3 Ds"—**dermatitis, dementia, and diarrhea**—is characteristic of **pellagra**, a severe deficiency of **niacin (vitamin B3)**. - **Cognitive decline** is a common manifestation of the neurological symptoms associated with dementia in pellagra. *Thiamine* - **Thiamine (vitamin B1)** deficiency causes **beriberi**, leading to cardiovascular (wet beriberi) or neurological (dry beriberi/Wernicke-Korsakoff syndrome) symptoms. - While **fatigue and cognitive impairment** can occur, the constellation of prominent dermatitis and severe dementia is not typical of primary thiamine deficiency. *Tryptophan* - **Tryptophan** is an **essential amino acid** and a precursor to niacin. A deficiency in tryptophan could indirectly lead to niacin deficiency. - However, the direct deficiency of tryptophan as the primary cause of the "3 Ds" is less common than a direct niacin deficiency or impaired niacin synthesis. *Retinol* - **Retinol (vitamin A)** deficiency primarily affects **vision**, leading to **night blindness** and xerophthalmia. - It also plays a role in immune function and epithelial cell integrity, but it does not cause the specific triad of dermatitis, dementia, and cognitive decline seen here. *Riboflavin* - **Riboflavin (vitamin B2)** deficiency causes **angular stomatitis, cheilosis, and glossitis** along with seborrheic dermatitis. - While it can cause skin manifestations, it does not typically present with the severe dementia and cognitive decline characteristic of pellagra.
Question 6: A 5-year-old girl was washing her doll with shampoo containing rotenone. Her mother noticed her in an unconscious state. Which enzyme is inhibited by the above chemical?
- A. NADH dehydrogenase (Correct Answer)
- B. Succinate dehydrogenase
- C. Cytochrome C
- D. Cytochrome oxidase
- E. Cytochrome b-c1 complex
Explanation: ***NADH dehydrogenase*** - **Rotenone** is a potent **inhibitor of mitochondrial complex I (NADH dehydrogenase)**, preventing the transfer of electrons from NADH to ubiquinone. - This inhibition disrupts the **electron transport chain**, leading to a halt in ATP synthesis and cellular energy failure, causing symptoms like unconsciousness. *Succinate dehydrogenase* - **Succinate dehydrogenase** (Complex II) is involved in both the **Krebs cycle and electron transport chain**, but is not directly inhibited by rotenone. - Only severe compromise of the electron transport chain can cause a secondary effect, but not direct enzyme inhibition. *Cytochrome C* - **Cytochrome C** is a mobile electron carrier in the electron transport chain, but it is not directly inhibited by rotenone. - **Cytochrome C** transfers electrons from complex III to complex IV. *Cytochrome oxidase* - **Cytochrome oxidase** (Complex IV) is responsible for the final transfer of electrons to oxygen, which is not inhibited by rotenone. - Inhibitors like **cyanide and carbon monoxide** specifically target **cytochrome oxidase**. *Cytochrome b-c1 complex* - **Cytochrome b-c1 complex** (Complex III) catalyzes electron transfer from ubiquinol to cytochrome C, but is not inhibited by rotenone. - This complex is specifically inhibited by **antimycin A**, not rotenone.
Question 7: A patient with high triglycerides (TG) esterified with long-chain fatty acids (LCFA) presents with fatigue, and a biopsy of the muscle shows fat vacuoles. What is the most likely diagnosis?
- A. Carnitine deficiency (Correct Answer)
- B. Fatty acid synthase defect
- C. Lipoprotein lipase (LPL) defect
- D. LDL defect
- E. Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency
Explanation: ***Carnitine deficiency*** - **Carnitine** is essential for transporting **long-chain fatty acids (LCFAs)** into the mitochondria for beta-oxidation. - A deficiency leads to the accumulation of **LCFAs** as **triglycerides** in the cytoplasm, resulting in **fat vacuoles** in muscle and systemic fatigue due to impaired energy production. *Fatty acid synthase defect* - **Fatty acid synthase** is involved in the *de novo* synthesis of fatty acids, not their catabolism or transport. - A defect would impair fatty acid production, not lead to the accumulation of **triglycerides** from exogenous sources. *Lipoprotein lipase (LPL) defect* - **LPL** is crucial for cleaving **triglycerides** in circulating chylomicrons and VLDL, allowing fatty acids to be taken up by tissues. - A defect causes severe hypertriglyceridemia, but the primary issue in the muscle with fat vacuoles points towards a problem with intracellular fatty acid utilization rather than plasma triglyceride clearance. *LDL defect* - **LDL** is primarily responsible for transporting cholesterol to peripheral tissues. - Defects in **LDL** metabolism typically lead to hypercholesterolemia, not the accumulation of **triglycerides** or muscle fat vacuoles as described. *Medium-chain acyl-CoA dehydrogenase (MCAD) deficiency* - **MCAD** deficiency is a fatty acid oxidation disorder affecting **medium-chain fatty acids** (C6-C12), not the **long-chain fatty acids** specifically mentioned in the stem. - It typically presents with hypoketotic hypoglycemia during fasting, often in infancy or childhood, rather than the characteristic muscle fat vacuole accumulation pattern seen with **carnitine deficiency**.
Question 8: A patient presents with xanthomas on the Achilles tendon. What is the most likely diagnosis?
- A. Familial hypercholesterolemia (Correct Answer)
- B. Tangier's disease
- C. Familial hyperchylomicronemia
- D. Familial dysbetalipoproteinemia
- E. Familial combined hyperlipidemia
Explanation: ***Familial hypercholesterolemia*** - **Xanthomas** on the **Achilles tendon** are a classic clinical sign of familial hypercholesterolemia, along with significantly elevated **LDL-C levels**. - This condition is an **autosomal dominant** genetic disorder characterized by defects in the **LDL receptor** pathway, leading to impaired clearance of LDL from the blood. - **Tendon xanthomas** (especially Achilles and extensor tendons) are pathognomonic for this condition. *Tangier's disease* - Characterized by very low or absent **HDL-C (high-density lipoprotein cholesterol)** levels, leading to **cholesterol ester accumulation** in various tissues. - While it can cause lipid deposition, its hallmark is **enlarged, orange tonsils** and peripheral neuropathy, not typically Achilles tendon xanthomas. *Familial hyperchylomicronemia* - This disorder primarily involves elevated **chylomicrons** and **triglycerides**, presenting with **eruptive xanthomas** (small, red-yellow papules) but not typically tendon xanthomas. - It is often associated with **pancreatitis** and **lipemia retinalis**. *Familial dysbetalipoproteinemia* - Characterized by elevated levels of **cholesterol** and **triglycerides** due to accumulation of remnant lipoproteins (IDL). - While it can cause **xanthomas**, these are typically **palmar xanthomas** (xanthoma striata palmaris) and **tuberoeruptive xanthomas**, less commonly Achilles tendon xanthomas. *Familial combined hyperlipidemia* - Most common familial lipid disorder, characterized by elevated **LDL-C** and/or **triglycerides** with variable phenotype. - While it causes premature coronary artery disease, it typically does **not** cause tendon xanthomas, which distinguishes it from familial hypercholesterolemia. - Xanthomas, if present, are usually **xanthelasma** (around eyelids) rather than tendon xanthomas.
Question 9: What is the source of Vitamin B1 (Thiamine)?
- A. A
- B. B
- C. C (Correct Answer)
- D. D
- E. E
Explanation: ***C*** - Element C appears to be a **grain-based flatbread**, which is a rich source of **thiamine (Vitamin B1)**. Whole grains, fortified cereals, and legumes are primary dietary sources of this essential vitamin. - **Thiamine** plays a crucial role in **carbohydrate metabolism** and nerve function. *A* - Element A appears to be **rice**, which is a **grain**. However, the question asks for the best source, and while rice can contain thiamine, the other option is a stronger candidate. - White rice, in particular, is often **enriched with thiamine** but naturally contains less than whole grains. *B* - Element B contains what appears to be **vegetables and salad**. While vegetables contain some thiamine, they are not typically considered the richest sources. - **Fruits and vegetables** are more commonly known for vitamins like C and A, and various minerals. *D* - Element D appears to be a **curry or stew**, likely containing lentils or other legumes. While some curries can contain thiamine, based on the image's clarity, it is not the most distinctly represented rich source compared to C. - The thiamine content in stews or curries can vary greatly depending on the **specific ingredients** used. *E* - Element E appears to be another food item in the image. While it may contain some nutrients, it is not the primary source of thiamine (Vitamin B1) among the options shown. - The **grain-based flatbread (C)** remains the richest and most reliable source of thiamine in this selection.
Question 10: A patient has multiple tendon xanthomas. Serum cholesterol ( $398 \mathrm{mg} / \mathrm{dL}$ ) and LDL ( 220 $\mathrm{mg} / \mathrm{dL}$ ) were found to be elevated. What is the most likely defect?
- A. Lipoprotein lipase deficiency
- B. LDL receptor defect (Correct Answer)
- C. Apo E defect
- D. LCAT deficiency
- E. Apo B-100 defect
Explanation: ***LDL receptor defect*** - **Tendon xanthomas** are a classic sign of **familial hypercholesterolemia**, which is most commonly caused by a genetic defect in the **LDL receptor**. - **Elevated LDL cholesterol** levels are a hallmark of this condition, as dysfunctional LDL receptors lead to impaired clearance of LDL particles from the blood. *Lipoprotein lipase deficiency* - This condition primarily causes severe **hypertriglyceridemia** and can lead to **eruptive xanthomas**, but not typically tendon xanthomas. - While cholesterol levels might be elevated, the defining feature would be very high triglyceride levels, often exceeding 1000 mg/dL. *Apo E defect* - A defect in **ApoE** (specifically the **ApoE2/E2 genotype**) is associated with **familial dysbetalipoproteinemia** (Type III hyperlipoproteinemia). - This condition causes elevated remnants of chylomicrons and VLDL, leading to **palmar xanthomas**, but less commonly tendon xanthomas, and often presents with high triglyceride levels in addition to cholesterol. *Apo B-100 defect* - **Familial defective apoB-100** can present similarly to familial hypercholesterolemia with elevated LDL cholesterol. - However, this is much **rarer** than LDL receptor defects (affecting ~1:700 vs 1:250-500 for LDL receptor mutations). - The clinical presentation and lipid profile overlap significantly, but LDL receptor defects remain the most common cause of this clinical picture. *LCAT deficiency* - **Lecithin-cholesterol acyltransferase (LCAT)** deficiency leads to an accumulation of **unesterified cholesterol** in plasma and tissues. - This typically presents with **corneal opacities**, **hemolytic anemia**, and proteinuria, rather than predominantly tendon xanthomas and isolated severe LDL elevation.