
Genetic and Metabolic Disorders — MCQs

On this page
Which of the following is not a feature of Down syndrome?
Chloride level in sweat is used in the diagnosis of which disease?
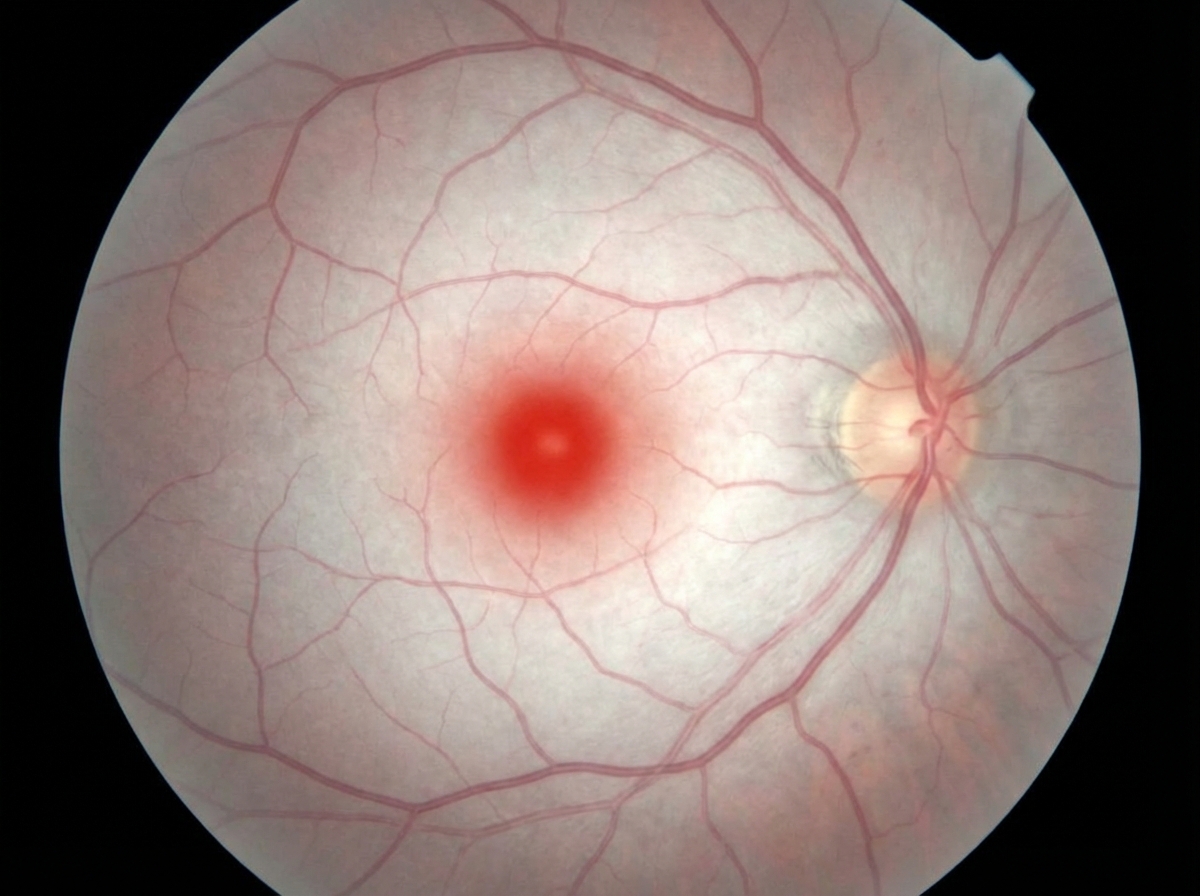
A 3-year-old boy presents with developmental delays and an inability to walk. The fundoscopy image is given below. What is the most likely diagnosis?
Risk of genetic diseases in consanguineous marriage between first cousins?
Which of the following statements about Fragile X syndrome is true?
Which of the following statements is MOST accurate regarding ataxia telangiectasia?
What is the current medical terminology used to refer to Down syndrome?
Alagille syndrome - which of the following statements is false?
Which of the following is a characteristic feature of DiGeorge syndrome?
Which of the following is NOT a feature of CHARGE syndrome?
Practice by Chapter
Chromosomal Disorders
Practice Questions
Disorders of Amino Acid Metabolism
Practice Questions
Disorders of Carbohydrate Metabolism
Practice Questions
Disorders of Lipid Metabolism
Practice Questions
Genetic Counseling
Practice Questions
Genetic Testing in Pediatrics
Practice Questions
Inborn Errors of Metabolism
Practice Questions
Lysosomal Storage Diseases
Practice Questions
Mitochondrial Disorders
Practice Questions
Multifactorial Inheritance Disorders
Practice Questions
Newborn Screening
Practice Questions
Single Gene Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app