
Genetic and Metabolic Disorders — MCQs

On this page
Acrodermatitis enteropathica is:-
An 8 year old boy complains of increasing muscle weakness. On examination, his calves are bulky and show muscle tightening. His serum creatine kinase levels are increasing with age. Which of the following is the most likely diagnosis?
A 7 year old boy with progressive muscle weakness and walking difficulties presented with the following finding. What is the probable diagnosis?

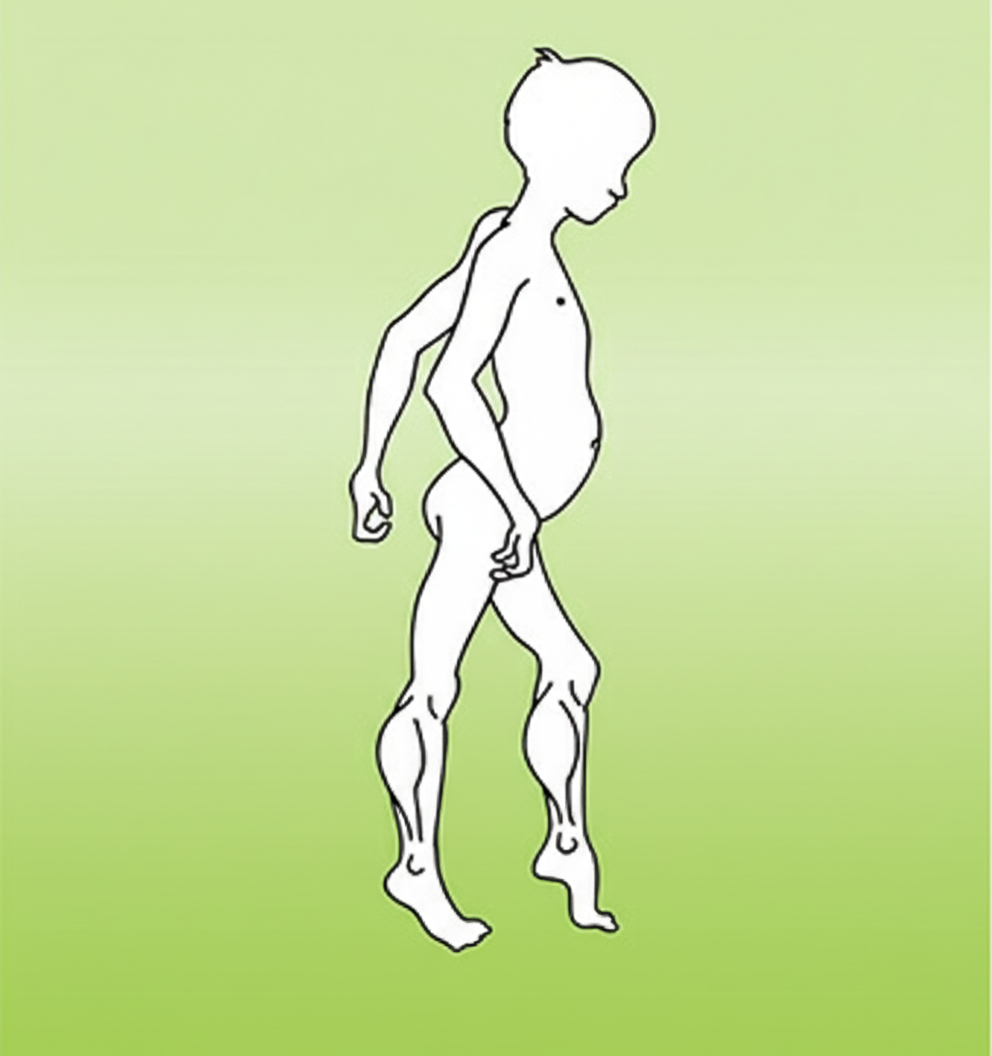
A 2-year- old boy presents with progressive clumsiness and difficulty walking. On physical examination, the child has large calves. He has difficulty walking on his toes and has a waddling gait as shown. Which of the following is the most likely diagnosis?
Ataxia telangiectasia is associated with all of the following except:
10-year old male with poor growth, poor appetite, recurrent chest infection and steatorrhea is likely to have ____________ .
Which of the following syndromes is associated with mental retardation?
A 7-year-old boy has demineralized bones with pseudofractures. Physiologic doses of vitamin D do not result in improvement. Which of the following is most likely to be associated with this syndrome?
4 year old male, recurrent URTI, has difficulty breathing, High arched palate, Failure to grow and impaired hearing, management is
Streak ovaries are seen in: CMC (Vellore) 09
Practice by Chapter
Chromosomal Disorders
Practice Questions
Disorders of Amino Acid Metabolism
Practice Questions
Disorders of Carbohydrate Metabolism
Practice Questions
Disorders of Lipid Metabolism
Practice Questions
Genetic Counseling
Practice Questions
Genetic Testing in Pediatrics
Practice Questions
Inborn Errors of Metabolism
Practice Questions
Lysosomal Storage Diseases
Practice Questions
Mitochondrial Disorders
Practice Questions
Multifactorial Inheritance Disorders
Practice Questions
Newborn Screening
Practice Questions
Single Gene Disorders
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app