
Congenital defects — MCQs

On this page
A 2600-g (5-lb 8-oz) male newborn is delivered at 34 weeks' gestation to a 22-year-old woman. The mother did not have prenatal care. Upon examination in the delivery room, the newborn's skin appears blue. He is gasping and breathing irregularly. The ears are low-set with broad auricles, and the nasal tip is flattened. The lower jaw is small and displaced backward. The right foot is clubbed. Which of the following is the most likely underlying cause of this patient's condition?
A newborn male is evaluated 30 minutes after birth. He was born at 38 weeks gestation to a 39-year-old gravida 3 via vaginal delivery. The pregnancy was complicated by gestational diabetes, and the patient’s mother received routine prenatal care. The family declined all prenatal testing, including an anatomy ultrasound. The patient’s two older siblings are both healthy. Upon delivery, the patient appeared well and had good respiratory effort. He was noted to have acrocyanosis, and his Apgar scores were 8 and 9 at one and five minutes of life, respectively. The patient’s birth weight is 3840 g (8 lb 7 oz). His temperature is 98.7°F (37.1°C), blood pressure is 66/37 mmHg, pulse is 142/min, and respirations are 34/min. On physical exam, the patient has low-set ears, upslanting palpebral fissures, and a hypoplastic fifth finger. Which of the following is most likely to be found in this patient?
A 3175-g (7-lb) female newborn is delivered at 37 weeks to a 26-year-old primigravid woman. Apgar scores are 8 and 9 at 1 and 5 minutes, respectively. The pregnancy had been uncomplicated. The mother had no prenatal care. She immigrated to the US from Brazil 2 years ago. Immunization records are not available. One day after delivery, the newborn's temperature is 37.5°C (99.5°F), pulse is 182/min, respirations are 60/min, and blood pressure is 82/60 mm Hg. The lungs are clear to auscultation. Cardiac examination shows a continuous heart murmur. The abdomen is soft and nontender. There are several discolored areas on the skin that are non-blanchable upon pressure application. Slit lamp examination shows cloudy lenses in both eyes. The newborn does not pass her auditory screening tests. Which of the following is the most likely diagnosis?
A female infant is born with a mutation in PKD1 on chromosome 16. An abdominal ultrasound performed shortly after birth would most likely reveal which of the following?
A previously healthy 2-year-old girl is brought to the physician because of a 1-week history of yellow discoloration of her skin, loss of appetite, and 3 episodes of vomiting. Her parents also report darkening of her urine and light stools. During the last 2 days, the girl has been scratching her abdomen and arms and has been crying excessively. She was born at 38 weeks' gestation after an uncomplicated pregnancy and delivery. Her family emigrated from Japan 8 years ago. Immunizations are up-to-date. Her vital signs are within normal limits. Examination shows jaundice of her skin and sclerae. Abdominal examination shows a mass in the right upper abdomen. Serum studies show: Bilirubin (total) 5 mg/dL Direct 4.2 mg/dL Aspartate aminotransferase (AST) 20 U/L Alanine aminotransferase (ALT) 40 U/L γ-Glutamyltransferase (GGT) 110 U/L Abdominal ultrasonography shows dilation of the gall bladder and a fusiform dilation of the extrahepatic bile duct. Which of the following is the most likely diagnosis?
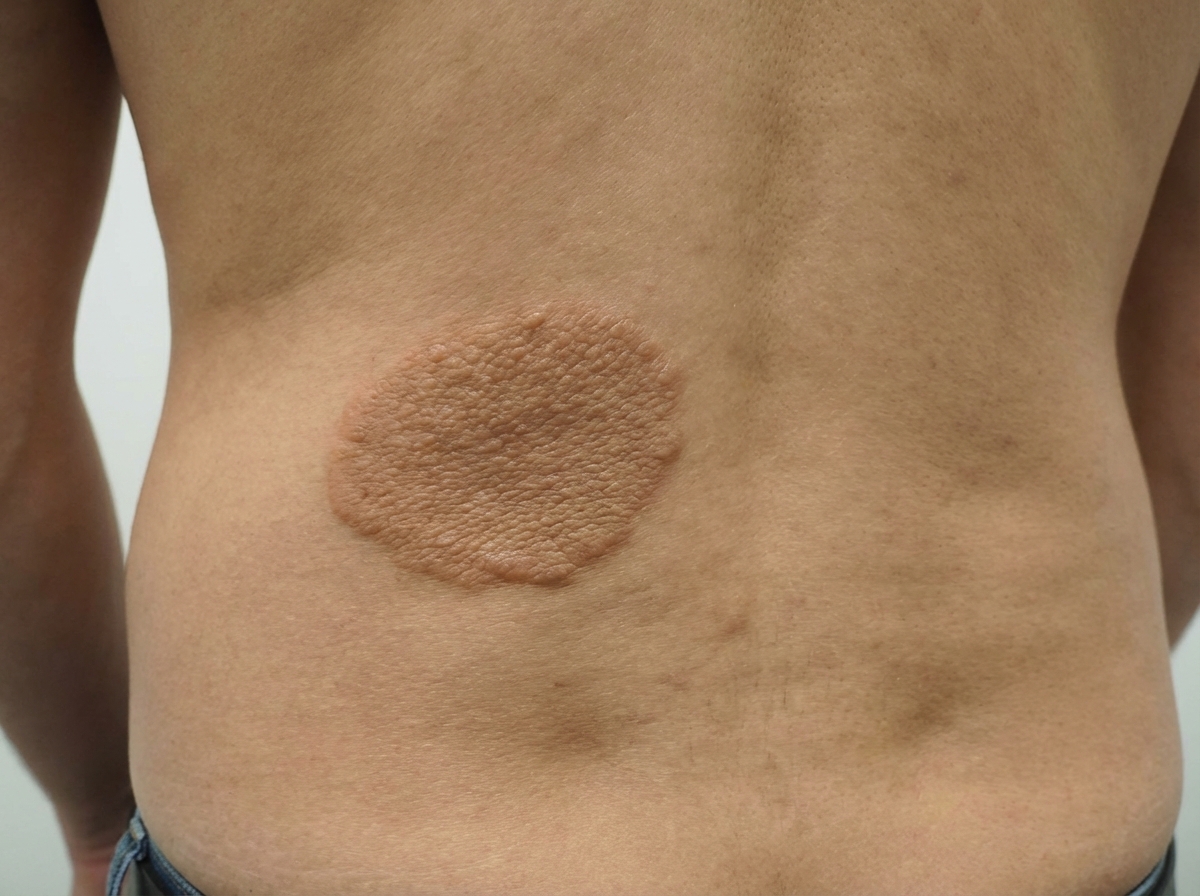
An 8-year-old boy presents with a skin lesion on his back as shown in the picture. On physical examination, there are synchronous spasmodic movements of the neck, trunk, and extremities. The physician explains that this is likely due to a genetic condition, and further testing would be necessary to confirm the diagnosis. Which of the following genes is involved in the development of this patient’s condition?
Cardiac surgery is consulted on a newborn with a large ventricular septal defect. The child has poor weight gain and feeding difficulties. He requires furosemide and captopril to avoid dyspnea. On physical examination his temperature is 36.9°C (98.4°F), pulse rate is 158/min, respiratory rate is 30/min, and blood pressure is 94/62 mm Hg. Chest auscultation reveals a holosystolic murmur along the left lower sternal border and a mid-diastolic low-pitched rumble at the apex. Abdominal examination reveals the presence of hepatomegaly. An echocardiogram confirms a diagnosis of a membranous VSD while hemodynamic studies show a Qp:Qs ratio of 2.8:1. Which of the following is the best management option?
A 3-week-old firstborn baby girl is brought to the pediatric emergency room with projectile vomiting. She started vomiting while feeding 12 hours ago and has been unable to keep anything down since then. After vomiting, she appears well and hungry, attempting to feed again. The vomitus has been non-bloody and non-bilious. The last wet diaper was 10 hours ago. The child was born at 40 weeks gestation to a healthy mother. On examination, the child appears sleepy but has a healthy cry during the exam. The child has dry mucous membranes and delayed capillary refill. There is a palpable olive-shaped epigastric mass on palpation. Which of the following is the most likely cause of this patient's condition?
A 28-year-old woman gives birth to a male infant. During her third-trimester antenatal sonogram, the radiologist noted a suspected congenital heart defect, but the exact nature of the defect was not clear. The pediatrician orders an echocardiogram after making sure that the baby’s vital signs are stable. This reveals the following findings: atresia of the muscular tricuspid valve, pulmonary outflow tract obstruction, open patent ductus arteriosus, a small ventricular septal defect, and normally related great arteries. The pediatrician explains the nature of the congenital heart defect to the infant's parents. He also informs them about the probable clinical features that are likely to develop in the infant, the proposed management plan, and the prognosis. Which of the following signs is most likely to manifest first in this infant?
A 3-week-old boy has non-bilious projectile vomiting that occurred after feeding. After vomiting, the infant is still hungry. The infant appears dehydrated and malnourished. A firm, “olive-like” mass of about 1.5 cm in diameter is palpated in the right upper quadrant, by the lateral edge of the rectus abdominus muscle. On laboratory testing, the infant is found to have a hypochloremic, hypokalemic metabolic alkalosis. Which of the following is most likely the cause of this patient’s symptoms?
Practice by Chapter
Neural tube defects
Practice Questions
Congenital heart defects
Practice Questions
Gastrointestinal malformations
Practice Questions
Genitourinary anomalies
Practice Questions
Craniofacial anomalies
Practice Questions
Skeletal dysplasias
Practice Questions
Chromosomal disorders
Practice Questions
Teratogenic exposures
Practice Questions
Multiple malformation syndromes
Practice Questions
Prenatal diagnosis of congenital defects
Practice Questions
Surgical management timing
Practice Questions
Long-term outcomes and follow-up
Practice Questions
Preventive strategies
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app