
Congenital defects — MCQs

On this page
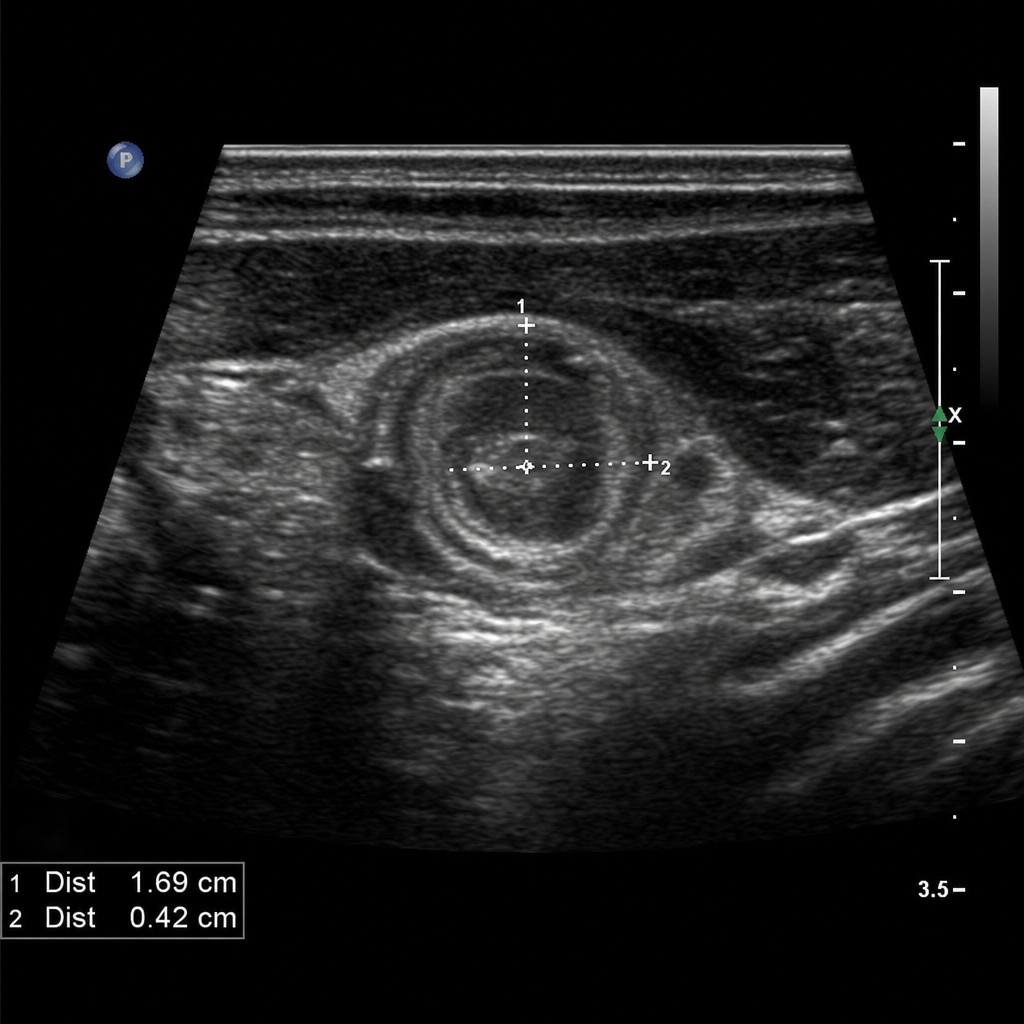
A 4-week-old male infant is brought to the emergency department with a 3-day history of progressive non-bilious, forceful vomiting after every feeding. He appears hungry and roots immediately after vomiting. On examination, weight is 3.1 kg (birth weight 3.4 kg), he is mildly lethargic, his anterior fontanelle is slightly sunken, and a small olive-shaped mass is palpable in the epigastrium. An abdominal ultrasound confirms the suspected diagnosis. Laboratory values show: Na 132 mEq/L, K 3.0 mEq/L, Cl 88 mEq/L, HCO3 30 mEq/L, pH 7.50. Which of the following represents the most appropriate sequence of management steps?
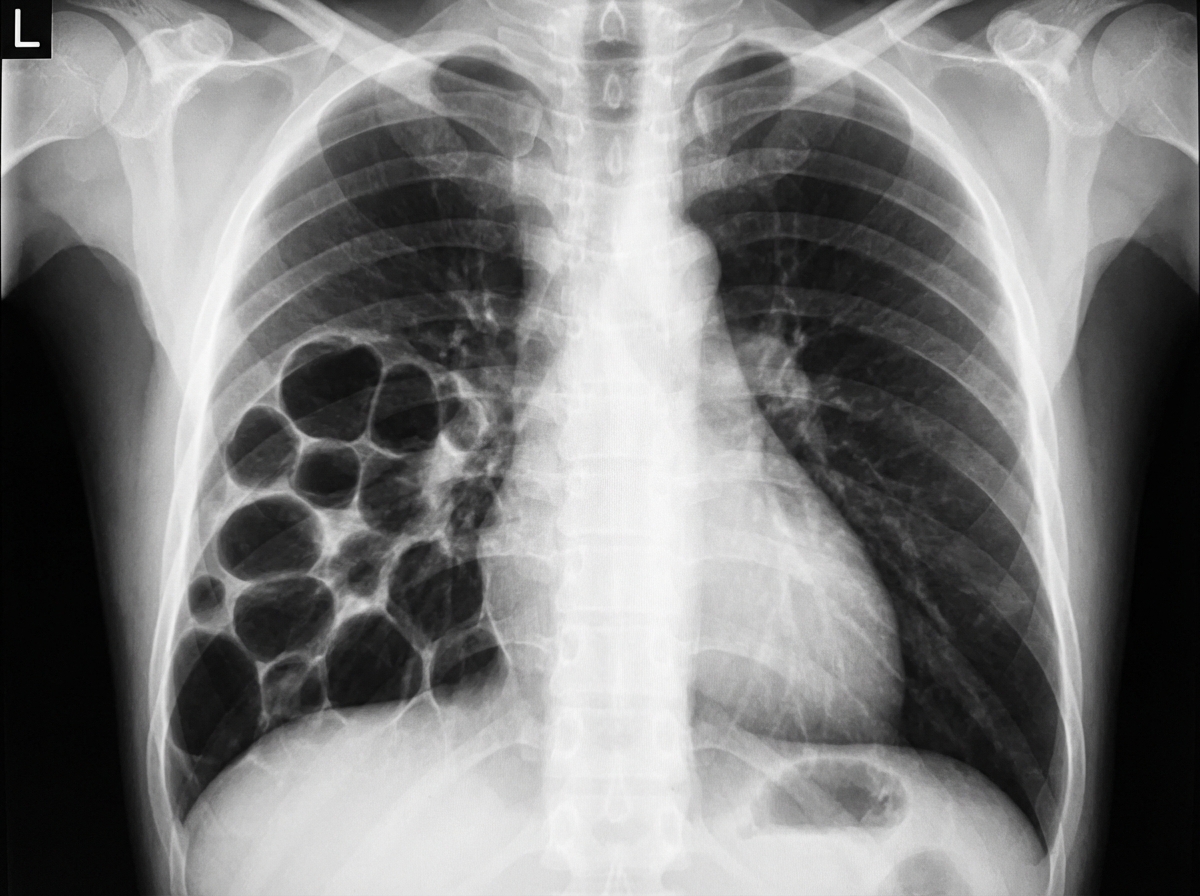
A 3-year-old child presents with respiratory distress and a history of recurrent respiratory infections. Based on the provided imaging, what is the most likely diagnosis?
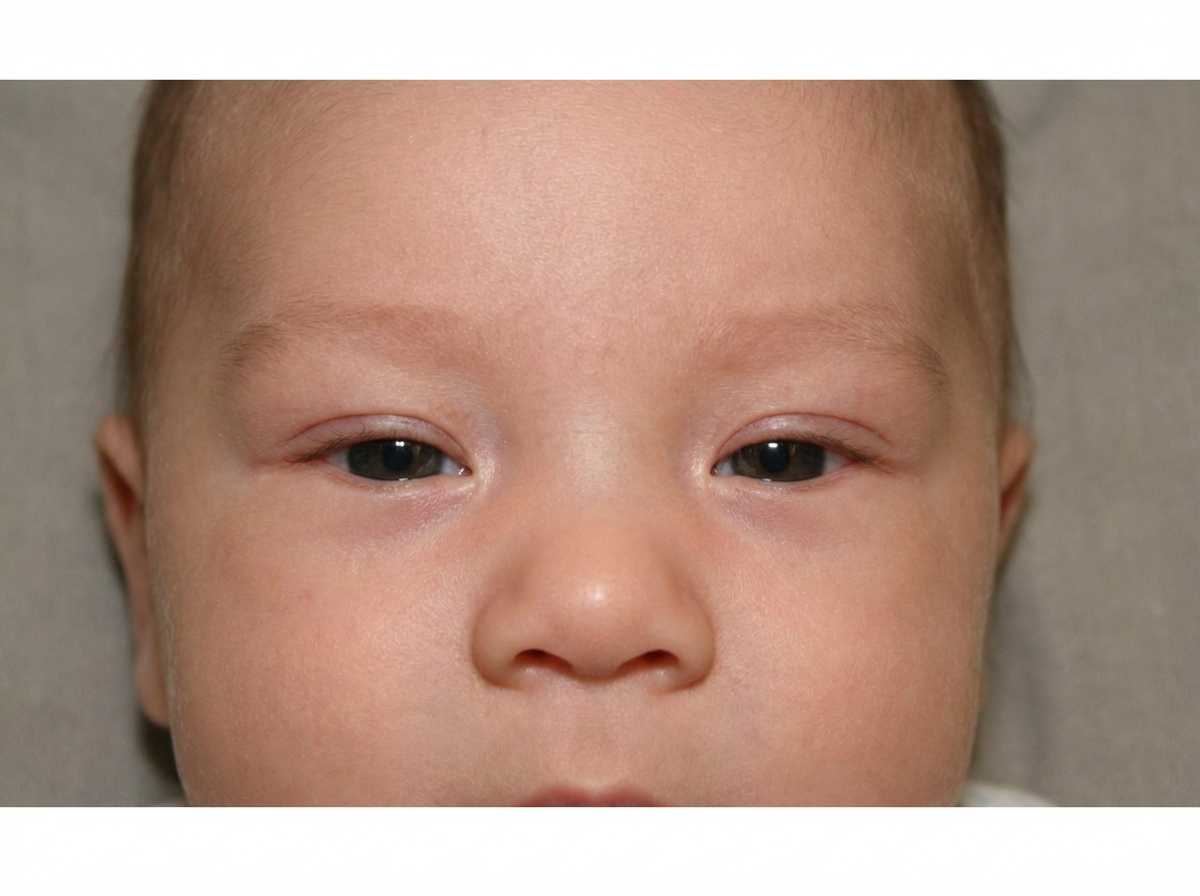
A 2 -month-old child presents with the following condition as shown in the image. What is the ideal management protocol?
A 13-year-old boy presents with jaundice, fatigue, muscle stiffness, tremors, and behavioral changes. Examination reveals an enlarged liver and spleen. A Kayser-Fleischer ring was noted. What is the definitive diagnostic test?
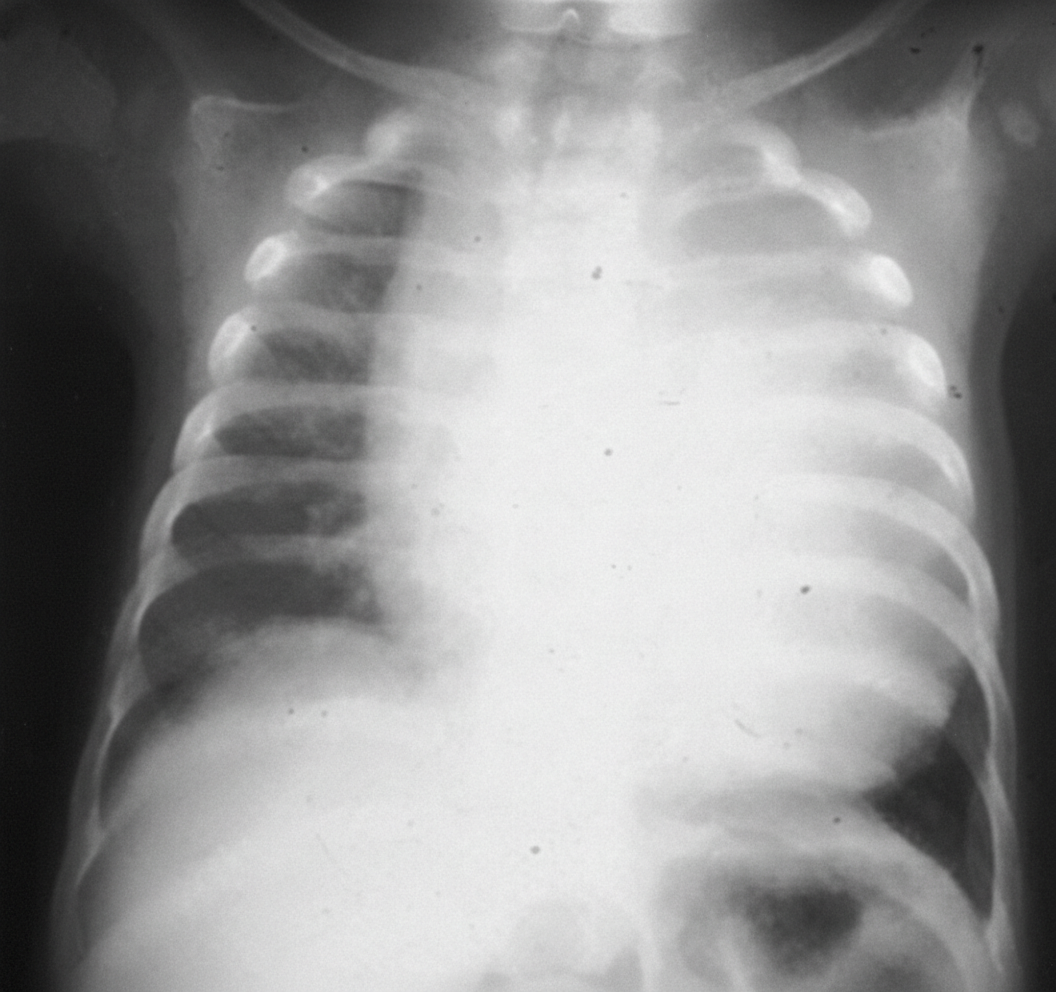
A patient presents with an X-ray showing cardiomegaly, along with symptoms of hypotonia, macroglossia, hepatomegaly, and floppy baby syndrome. The X ray of the infant is shown below. What is the most likely diagnosis?
Practice by Chapter
Neural tube defects
Practice Questions
Congenital heart defects
Practice Questions
Gastrointestinal malformations
Practice Questions
Genitourinary anomalies
Practice Questions
Craniofacial anomalies
Practice Questions
Skeletal dysplasias
Practice Questions
Chromosomal disorders
Practice Questions
Teratogenic exposures
Practice Questions
Multiple malformation syndromes
Practice Questions
Prenatal diagnosis of congenital defects
Practice Questions
Surgical management timing
Practice Questions
Long-term outcomes and follow-up
Practice Questions
Preventive strategies
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app