
Neuropathology — MCQs

On this page
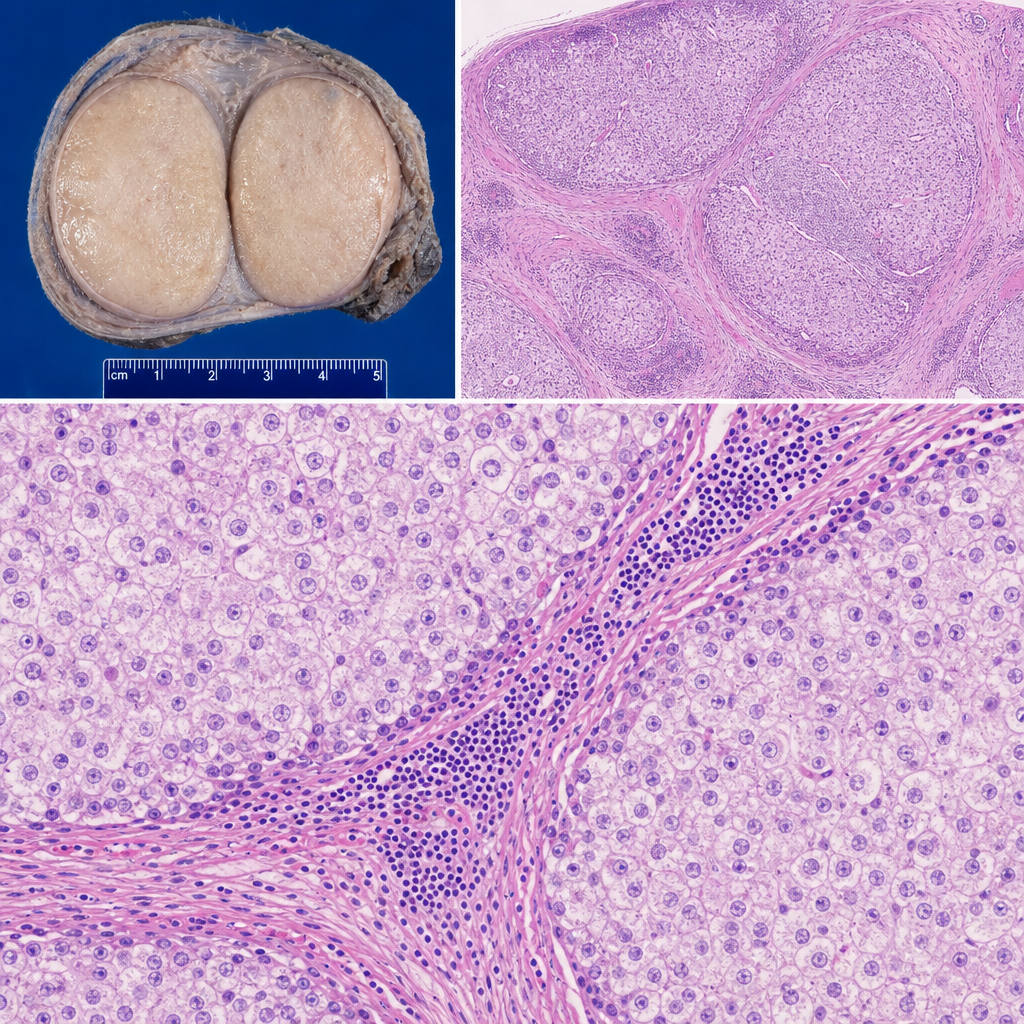
A 28-year-old man presents with a painless right testicular mass. Orchiectomy is performed. Gross examination reveals a 4 cm well-circumscribed, homogeneous, pale tan-gray mass without hemorrhage or necrosis. Histological sections show a monotonous population of large cells with clear, glycogen-rich cytoplasm and central round nuclei with prominent nucleoli, arranged in nests separated by fibrous septa infiltrated by mature lymphocytes. Which of the following best explains why the lymphocytic infiltrate is present within the fibrous septa of this tumor?
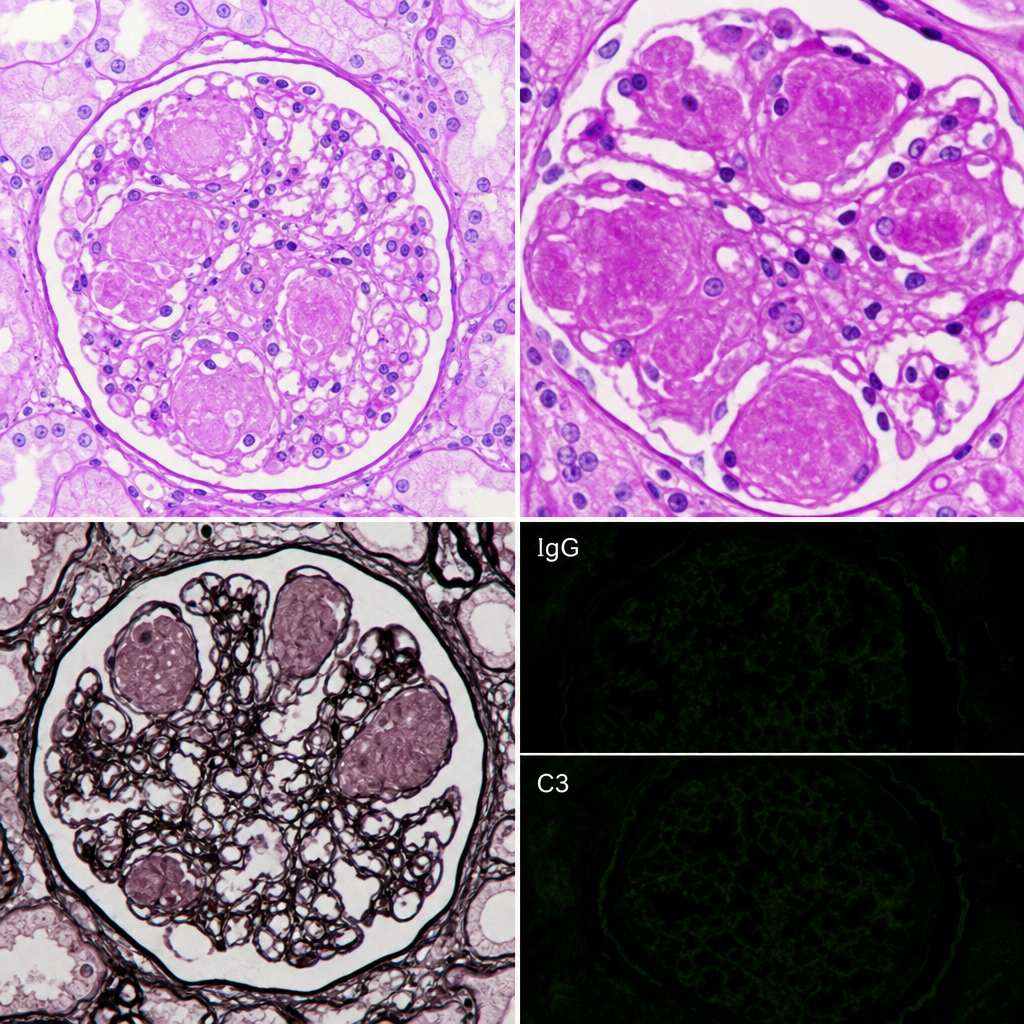
A 62-year-old man with poorly controlled type 2 diabetes mellitus for 18 years presents with proteinuria of 4.2 g/day and a serum creatinine of 2.8 mg/dL. Renal biopsy is performed. The biopsy shows PAS-positive, homogeneous, acellular nodular deposits in the mesangium of glomeruli, with associated capillary loop obliteration and diffuse glomerular basement membrane thickening. Immunofluorescence is negative for immune complex deposits and monoclonal light chain staining. Which of the following pathological processes best explains the formation of the nodular deposits seen in this biopsy?
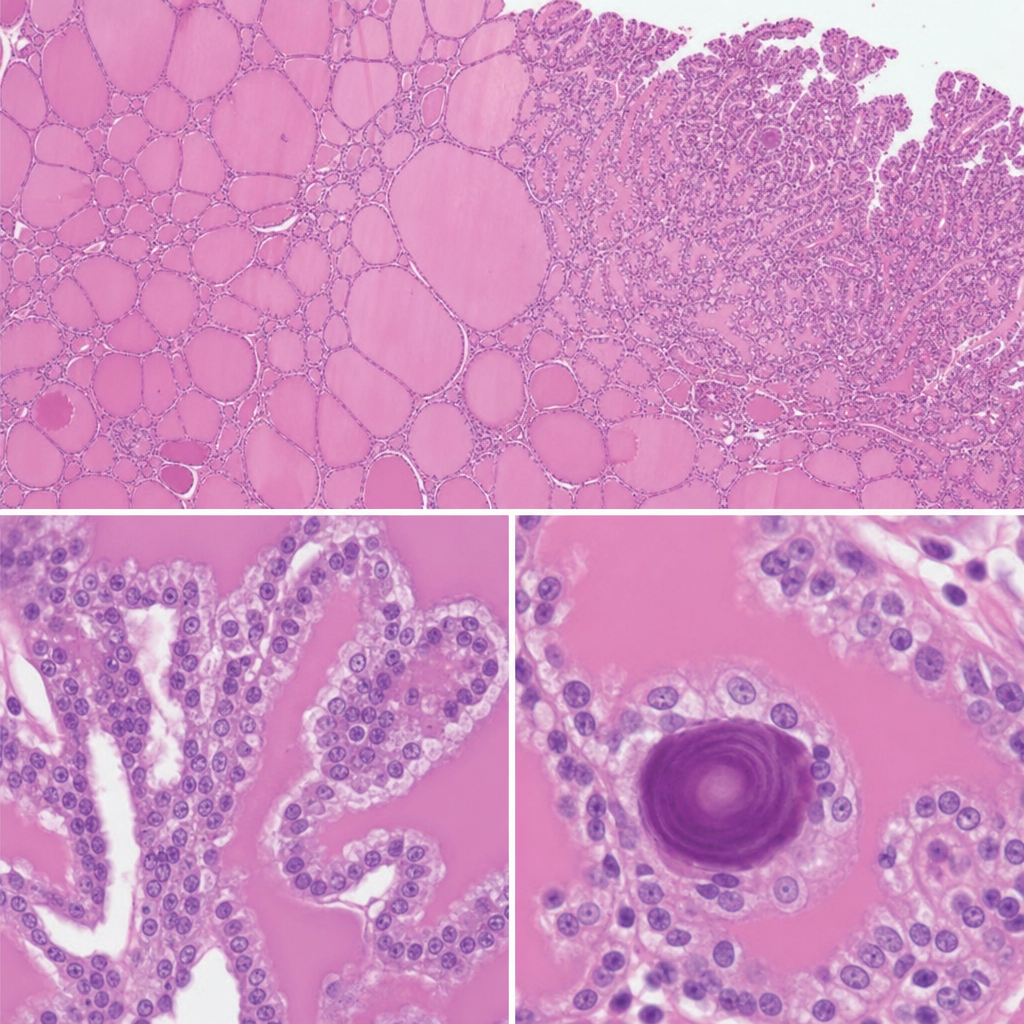
A 34-year-old woman undergoes excision of a neck mass. Histological examination of the resected specimen shows follicular structures of varying size filled with colloid, lined by cuboidal epithelium, with focal areas where small papillary projections bearing cells with ground-glass nuclei and nuclear grooves project into the follicular lumen. Scattered concentrically laminated calcified spherules are also identified within the papillary cores. Which of the following is the most likely origin and behavior of this neoplasm?
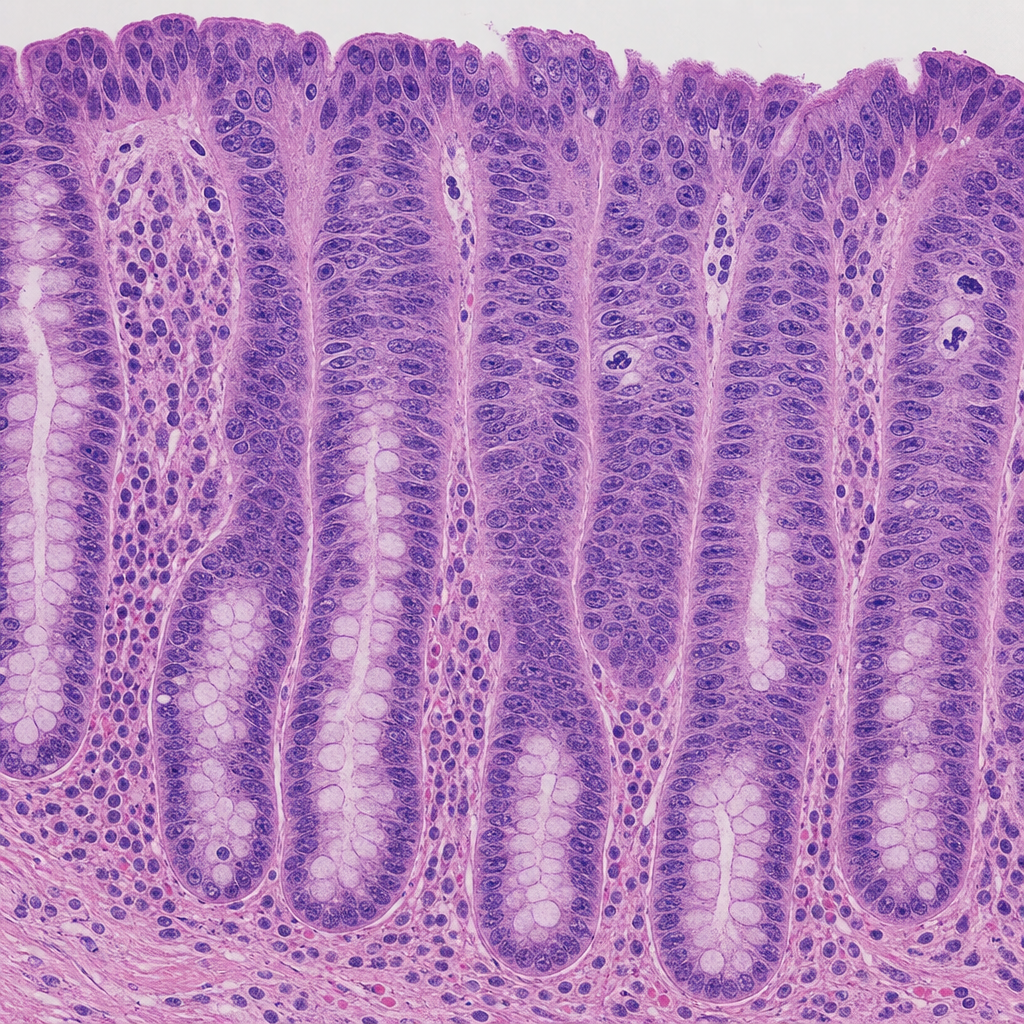
A 52-year-old man with a 25-year history of ulcerative colitis undergoes surveillance colonoscopy. A flat, velvety mucosal lesion is identified in the sigmoid colon and biopsied. The photomicrograph shows colonic crypts lined by cells with enlarged, hyperchromatic, stratified nuclei that extend to the luminal surface; the nuclei-to-cytoplasm ratio is markedly increased; goblet cells are absent; and mitotic figures including atypical forms are present in the upper third of the crypts. The basement membrane is intact with no stromal desmoplasia. Which of the following most accurately characterizes this lesion according to WHO classification principles?
A 35-year-old woman presents with headaches and seizures. MRI shows a well-circumscribed, calcified frontal lobe mass. Histology reveals oligodendroglioma with 1p/19q codeletion and IDH1 mutation. She undergoes gross total resection. Two years later, surveillance MRI shows a new enhancing nodule at the resection margin. Biopsy shows increased mitotic activity, microvascular proliferation, and retained 1p/19q codeletion but new CDKN2A/B homozygous deletion. What is the most critical factor in determining management strategy?
A 55-year-old man presents with progressive supranuclear gaze palsy, axial rigidity, and frequent falls. MRI shows midbrain atrophy with hummingbird sign. He dies 7 years later. Autopsy reveals globose neurofibrillary tangles in the basal ganglia and brainstem. Tau immunostaining shows 4-repeat tau predominance. His brother had similar symptoms. Genetic testing reveals a MAPT mutation. How does this change the pathogenic understanding and potential therapeutic approach?
A 70-year-old man with progressive dementia undergoes autopsy. Microscopy shows neuritic plaques and neurofibrillary tangles meeting criteria for Alzheimer disease (AD). However, sections also reveal Lewy bodies in the substantia nigra and cortex, moderate atherosclerosis with old lacunar infarcts, and TDP-43 positive inclusions in the hippocampus. He had no parkinsonian features clinically. What is the most appropriate neuropathologic interpretation?
A 42-year-old woman presents with behavioral changes, memory loss, and myoclonus. MRI shows cortical ribboning and T2 hyperintensity in the caudate and putamen. EEG shows periodic sharp wave complexes. CSF 14-3-3 is elevated, but real-time quaking-induced conversion (RT-QuIC) is negative. PRNP gene sequencing reveals E200K mutation. Her mother died of similar symptoms at age 45. What feature distinguishes this case from sporadic disease?
A 58-year-old man with HIV (CD4 count 45 cells/μL) presents with seizures and altered mental status. MRI shows multiple ring-enhancing lesions in the basal ganglia and cortex. Despite empiric treatment for toxoplasmosis for 2 weeks, lesions enlarge. Brain biopsy shows necrosis with surrounding large cells containing intranuclear inclusions and ground-glass nuclei. JC virus PCR is negative. What explains the unusual presentation and biopsy findings?
A 6-year-old boy presents with seizures and a calcified brain lesion on CT. Surgical resection shows a cystic tumor with a mural nodule. Histology reveals elongated bipolar cells with Rosenthal fibers and eosinophilic granular bodies. The tumor cells are GFAP-positive. Despite complete resection, which factor would most significantly impact long-term prognosis?
Practice by Chapter
Cerebrovascular diseases
Practice Questions
CNS trauma
Practice Questions
CNS infections
Practice Questions
Demyelinating diseases
Practice Questions
Neurodegenerative diseases
Practice Questions
Alzheimer's disease pathology
Practice Questions
Parkinson's disease pathology
Practice Questions
Amyotrophic lateral sclerosis
Practice Questions
Primary CNS tumors
Practice Questions
Metastatic CNS tumors
Practice Questions
Peripheral nerve disorders
Practice Questions
Skeletal muscle diseases
Practice Questions
Congenital CNS malformations
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app