
Immunopathology — MCQs

On this page
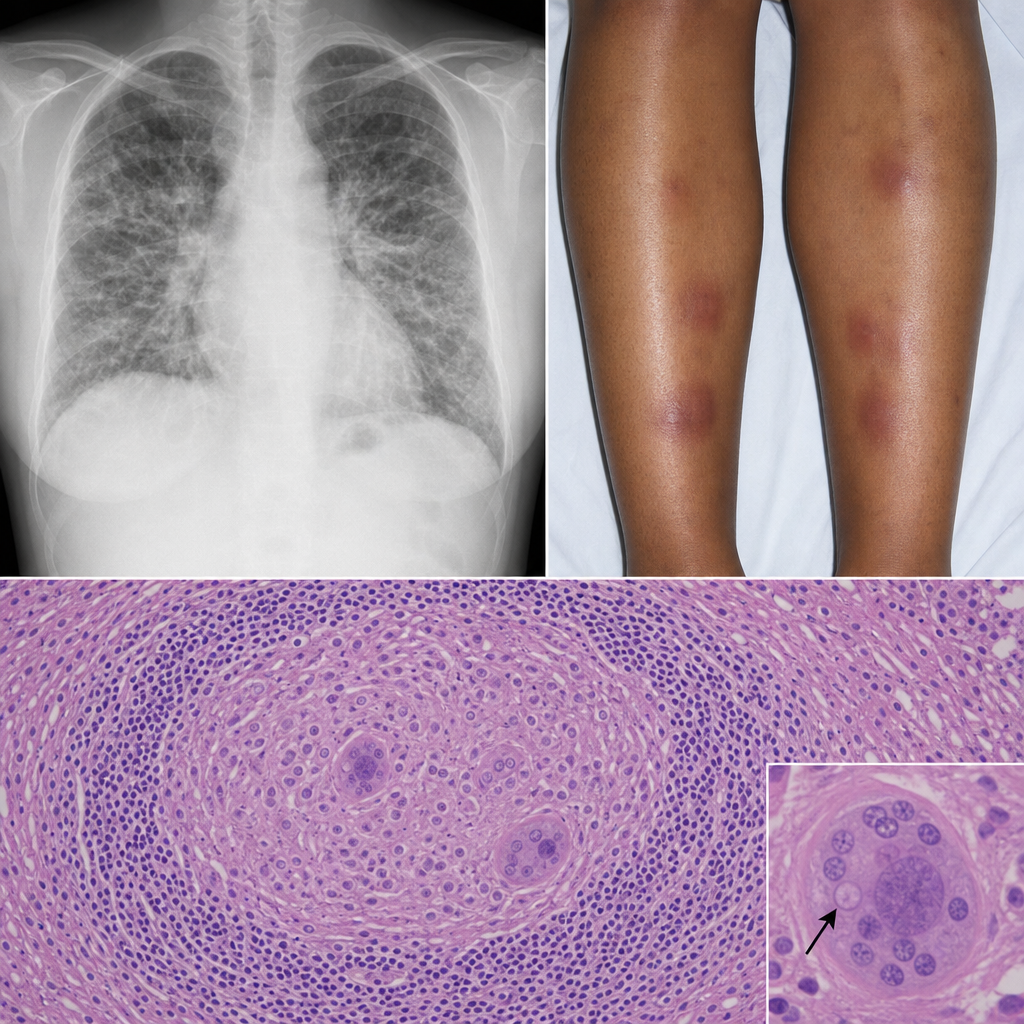
A 32-year-old Black woman with no occupational exposures presents with progressive dyspnea, fatigue, and bilateral ankle swelling. She also reports erythema nodosum on her shins. A chest radiograph shows bilateral hilar enlargement and diffuse interstitial infiltrates. Bronchoscopic biopsy is performed. The biopsy shows discrete, well-formed granulomas without central necrosis, surrounded by a rim of lymphocytes, with occasional Schaumann bodies and asteroid inclusions within giant cells. No organisms are identified on special stains. Which of the following best describes the predominant immunological mechanism driving this morphological pattern?
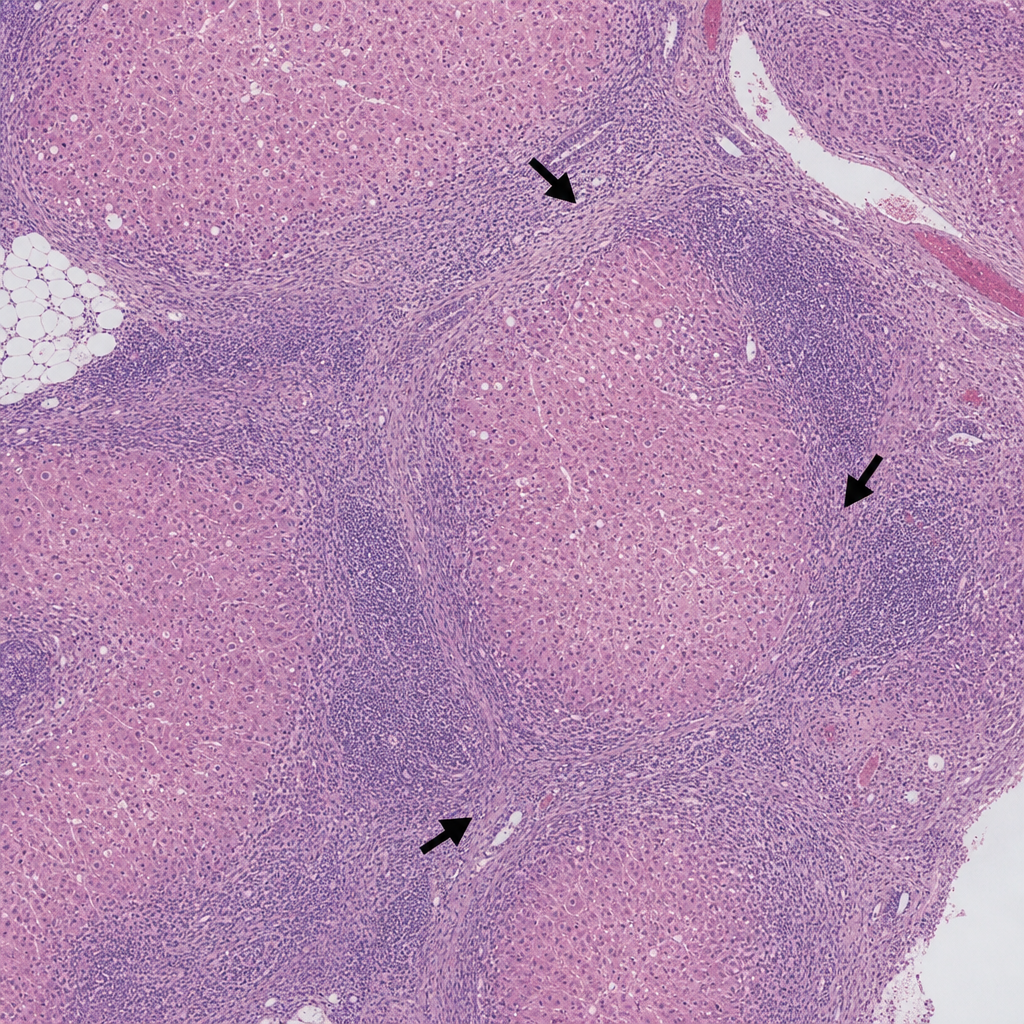
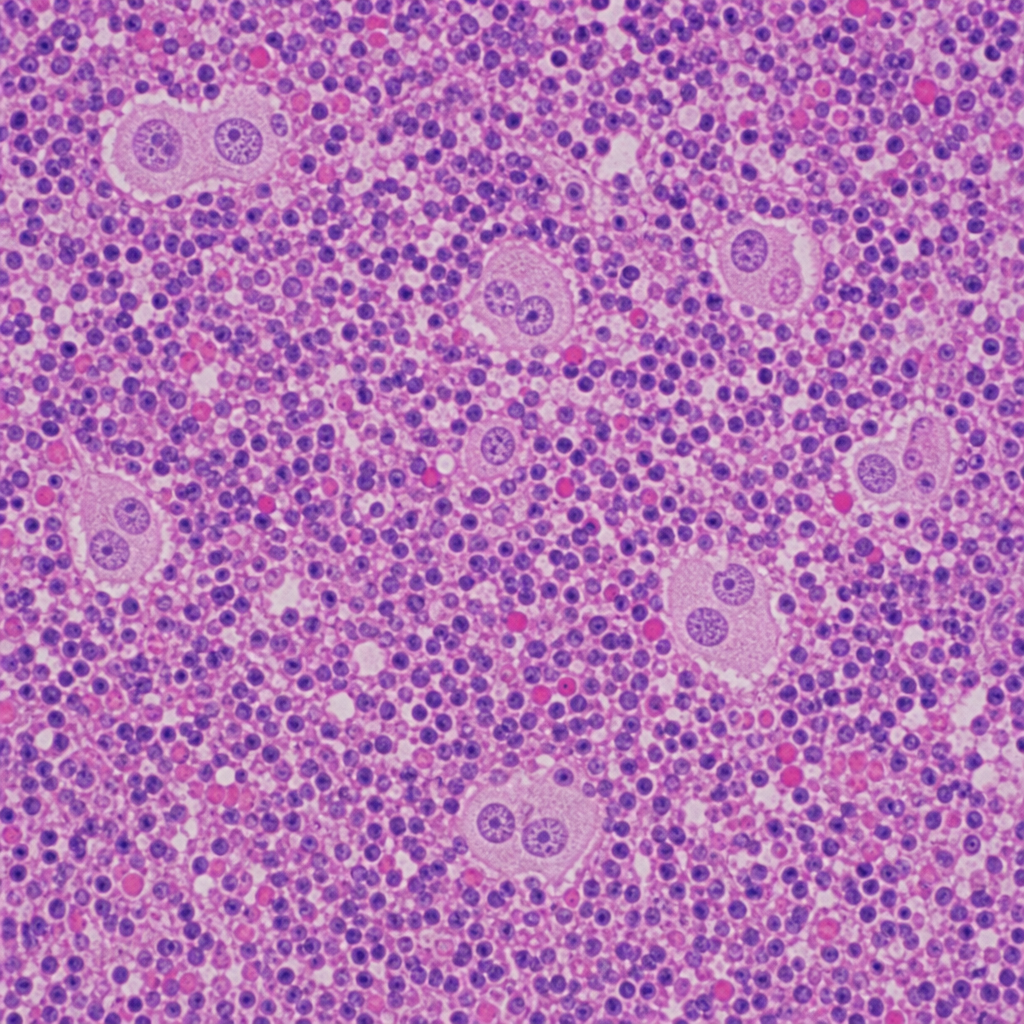
A 45-year-old woman presents with fatigue and jaundice. Serology is positive for anti-smooth muscle antibodies and elevated IgG. A liver biopsy is performed and shows a dense lymphoplasmacytic infiltrate expanding the portal tracts with interface hepatitis (piecemeal necrosis), plasma cell rosettes, and scattered acidophil bodies within the lobule. The lobular architecture is preserved. Which of the following findings, if present on the same biopsy, would most strongly support progression to an irreversible stage of this disease process?
A 34-year-old woman presents with painless cervical lymphadenopathy, night sweats, and a 6 kg weight loss over 3 months. Excisional lymph node biopsy is performed. The photomicrograph demonstrates effacement of nodal architecture by a mixed inflammatory infiltrate of lymphocytes, plasma cells, eosinophils, and neutrophils. Scattered large binucleated cells with prominent eosinophilic 'owl-eye' nucleoli are identified; these cells are CD15+, CD30+, CD20−, and CD45−. A second population of mononuclear variants with similar nuclear features is also present. Which of the following most accurately characterizes the large binucleated cells in this biopsy?
A 40-year-old woman with myasthenia gravis on pyridostigmine develops worsening weakness, diplopia, and dysphagia. She recently had URI and received azithromycin. Exam shows bilateral ptosis, ophthalmoplegia, and proximal muscle weakness with preserved reflexes. Her acetylcholinesterase inhibitor dose was increased 3 days ago. Edrophonium test shows no improvement. ABG shows hypercapnia. Evaluate the synthesis of clinical findings and determine the life-threatening complication requiring immediate intervention.
A 32-year-old pregnant woman at 20 weeks gestation with known anti-Rh(D) antibodies from previous pregnancy presents for routine prenatal care. Her current fetus is Rh(D)-positive by cell-free DNA testing. Middle cerebral artery Doppler shows increased peak systolic velocity. Fetal ultrasound reveals polyhydramnios and ascites. Amniocentesis shows elevated bilirubin. Synthesize the pathophysiology and evaluate the therapeutic intervention that addresses the underlying immune mechanism.
A 25-year-old man with HIV (CD4 count 450 cells/μL) on antiretroviral therapy for 6 months develops fever, lymphadenopathy, and worsening respiratory symptoms. Chest CT shows new mediastinal lymphadenopathy and pulmonary infiltrates. Sputum is positive for Mycobacterium tuberculosis. His viral load is undetectable. Evaluate the immunologic phenomenon responsible for his clinical deterioration despite virologic control.
A 55-year-old man with chronic hepatitis C develops palpable purpura on lower extremities, arthralgias, and weakness. Labs show elevated creatinine (2.5 mg/dL), low C4 (8 mg/dL), normal C3, positive rheumatoid factor, and cryoglobulins. Skin biopsy shows leukocytoclastic vasculitis. Renal biopsy reveals membranoproliferative glomerulonephritis with subendothelial deposits. Analyze the pathophysiologic link between the viral infection and systemic manifestations.
A 3-month-old infant presents with failure to thrive, chronic diarrhea, and oral candidiasis. Physical exam reveals absent tonsils and lymph nodes. Chest X-ray shows no thymic shadow. Flow cytometry reveals CD3+ T cells <200/μL, normal CD19+ B cells, and normal NK cells. Serum immunoglobulins are low despite normal B cell counts. Analyze the immunologic defect and predict the long-term complications.
A 42-year-old woman with Graves' disease treated with radioactive iodine develops periorbital edema, proptosis, and diplopia. TSH is suppressed, but free T4 is normal. Anti-TSH receptor antibodies are elevated. MRI shows enlarged extraocular muscles with increased retro-orbital fat. Analyze the immunopathologic mechanism causing her ophthalmopathy despite thyroid function normalization.
A 35-year-old woman receives a kidney transplant from her HLA-matched sibling. She is maintained on tacrolimus and mycophenolate. Three months post-transplant, her creatinine rises from 1.0 to 2.8 mg/dL over 2 weeks. Biopsy shows tubulitis with mononuclear cell infiltration and tubular epithelial damage. C4d staining is negative. Apply transplant immunology to determine the rejection mechanism.
Practice by Chapter
Hypersensitivity reactions (types I-IV)
Practice Questions
Autoimmune mechanisms
Practice Questions
Systemic autoimmune diseases
Practice Questions
Organ-specific autoimmune diseases
Practice Questions
Immunodeficiency disorders
Practice Questions
Primary immunodeficiencies
Practice Questions
Secondary immunodeficiencies
Practice Questions
AIDS pathology
Practice Questions
Amyloidosis types
Practice Questions
Transplant rejection mechanisms
Practice Questions
Graft-versus-host disease
Practice Questions
Immune-related adverse events
Practice Questions
Immune checkpoint pathways
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app