
Endocrine pathology — MCQs

On this page
A 28-year-old man presents with painless cervical lymphadenopathy. Excisional lymph node biopsy is performed. The photomicrograph shows effacement of normal nodal architecture by a mixed infiltrate of lymphocytes, plasma cells, eosinophils, and neutrophils. Scattered among the infiltrate are large binucleated cells with prominent eosinophilic 'owl-eye' nucleoli. Immunohistochemistry shows these large cells are CD15+, CD30+, CD20−, and CD45−. Which of the following cell types represents the malignant population in this lesion?
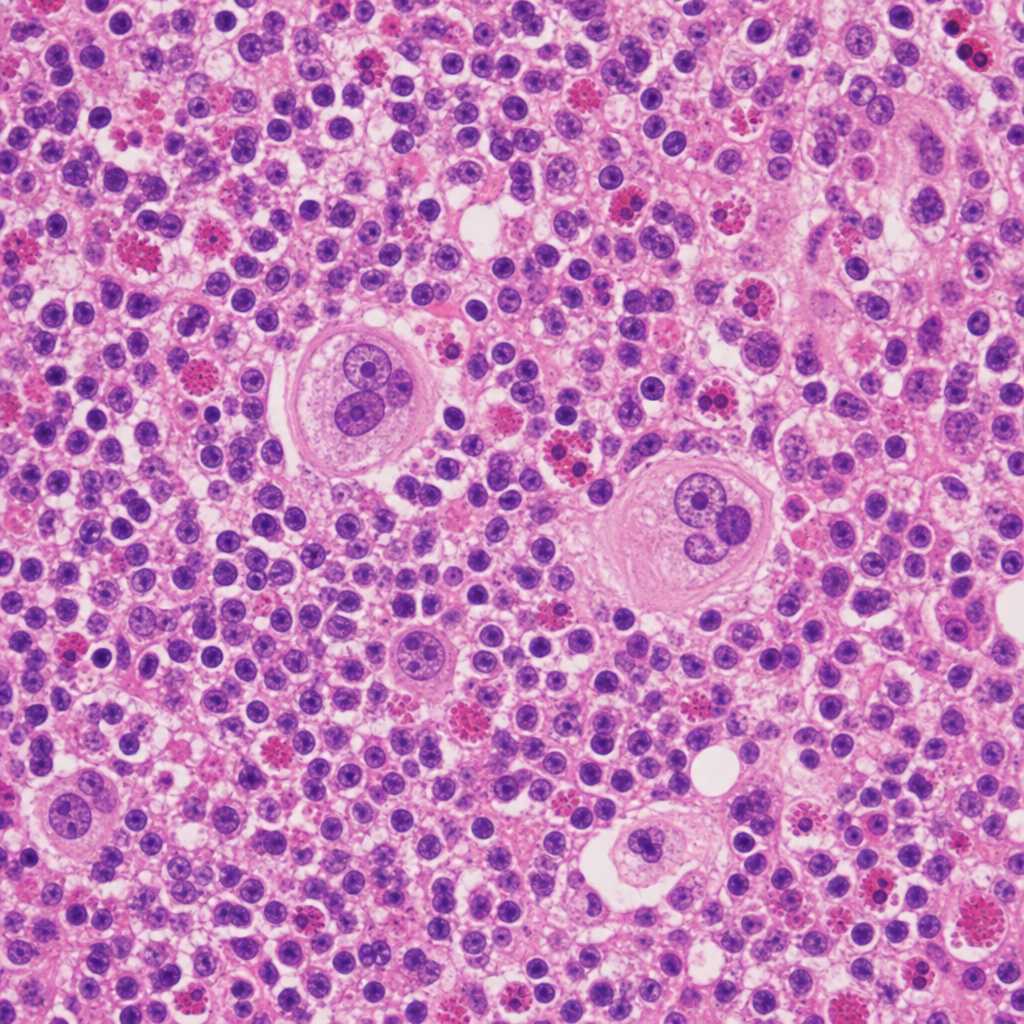
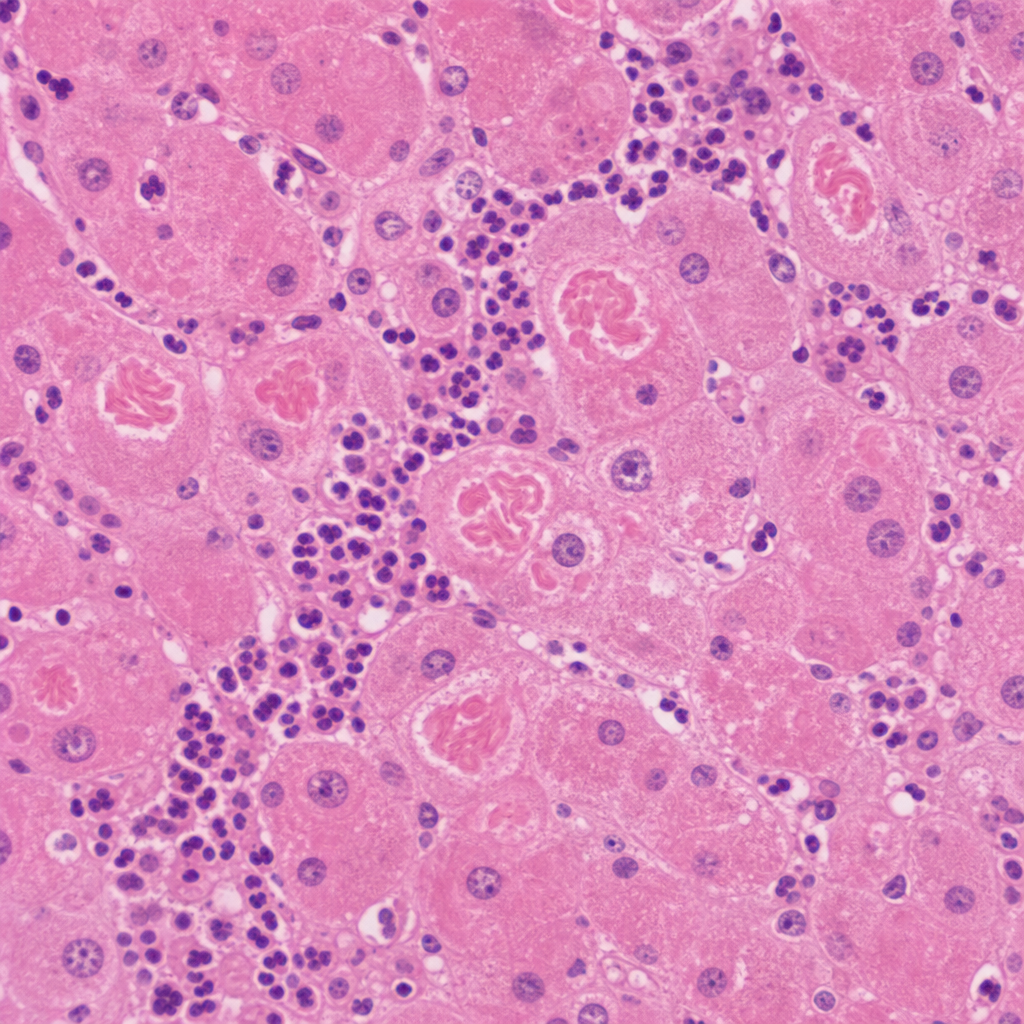
A 67-year-old man with a history of chronic alcohol use and cirrhosis undergoes liver biopsy for evaluation of worsening hepatic function. The photomicrograph shows hepatocytes with eosinophilic, irregularly shaped intracytoplasmic rope-like inclusions. Surrounding these cells is a neutrophilic infiltrate. The inclusions stain positively with ubiquitin immunohistochemistry. Which of the following best describes the composition of the inclusions seen in this biopsy?
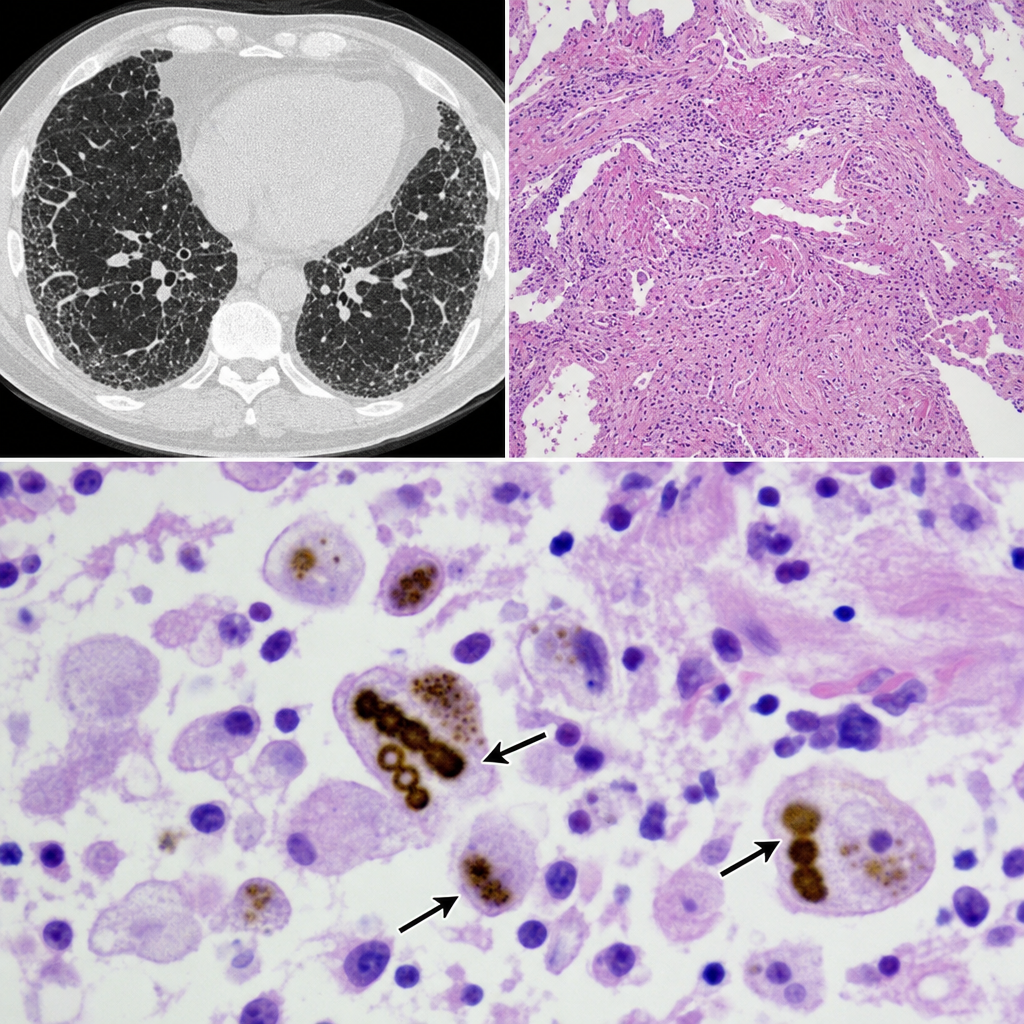
A 58-year-old man with a 40-pack-year smoking history presents with progressive dyspnea and a non-productive cough. Chest CT shows bilateral lower-lobe predominant interstitial thickening. An open lung biopsy is performed. The biopsy reveals golden-brown, beaded, dumbbell-shaped structures coated with iron-protein complexes within alveolar macrophages, accompanied by interstitial fibrosis. Which of the following is the most accurate characterization of these structures?
A 38-year-old woman with type 1 diabetes for 20 years presents with diabetic ketoacidosis. She is treated and recovers. Six months later, she develops progressive fatigue, nausea, and hyperpigmentation. Laboratory studies show morning cortisol 3 μg/dL, ACTH 180 pg/mL, TSH 8.2 mIU/L, free T4 0.6 ng/dL, and positive anti-thyroid peroxidase antibodies. She also has positive 21-hydroxylase antibodies. Her 12-year-old daughter was recently diagnosed with type 1 diabetes. Evaluate the pathologic process and most critical monitoring recommendation for the daughter.
A 29-year-old man presents with severe headaches and episodic palpitations, sweating, and anxiety. During an episode, blood pressure is 240/130 mmHg. Between episodes, blood pressure is 135/85 mmHg. 24-hour urine shows metanephrines 4.2 mg (normal: <1.0). MRI reveals a 4 cm right adrenal mass. His brother died suddenly at age 25 from an intracranial hemorrhage, and his father had thyroid cancer. Genetic testing reveals a RET proto-oncogene mutation. Evaluate the pathologic syndrome and preoperative management priority.
Practice by Chapter
Pituitary disorders
Practice Questions
Thyroid diseases (hyper/hypothyroidism)
Practice Questions
Thyroiditis
Practice Questions
Thyroid neoplasms
Practice Questions
Parathyroid disorders
Practice Questions
Adrenal cortical diseases
Practice Questions
Adrenal medullary disorders
Practice Questions
Pheochromocytoma and paraganglioma
Practice Questions
Pancreatic endocrine tumors
Practice Questions
Multiple endocrine neoplasia syndromes
Practice Questions
Diabetes mellitus pathology
Practice Questions
Endocrine disorders in pregnancy
Practice Questions
Endocrine effects of non-endocrine tumors
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app