
Urea cycle — MCQs

On this page
A 4-year-old boy presents with vomiting and one day of lethargy after a one week history of flu-like symptoms and low-grade fevers. The vomiting is nonbilious and nonbloody. The patient has had no other symptoms aside from mild rhinorrhea and cough. He has no past medical history, and is on no medications except for over-the-counter medications for his fever. His temperature is 98.5°F (36.9°C), pulse is 96/min, respirations are 14/min, and blood pressure is 108/80 mmHg. The patient appears lethargic and is oriented only to person. Otherwise, the physical exam is benign and the patient has no other neurologic symptoms. What is the mechanism of the most likely cause of this patient’s presentation?
A 3-year-old boy is seen in clinic. He was born at home without perinatal care. He was apparently normal at birth, but later developed failure to thrive and developmental delay. He also has a history of cataracts. His older brother had a myocardial infarction at the age of 18 and is rather lanky and tall in appearance. Laboratory testing of his urine showed an increase in the level of an amino acid. What is the most likely mechanism responsible for this boy's pathology?
A newborn boy develops projectile vomiting 48 hours after delivery. He is found to be lethargic, with poor muscle tone, and is hyperventilating. Within hours, he suffers important neurological deterioration, leading to seizures, coma, and, ultimately, death. An autopsy is performed and the pathology team makes a diagnosis of a rare genetic disorder that leads to low levels of N-acetylglutamate. Which of the following enzymes would be secondarily affected by this process?
A 3-week old boy is brought to the physician for the evaluation of lethargy, recurrent vomiting, and poor weight gain since birth. Physical examination shows decreased skin turgor and a bulging frontal fontanelle. Serum studies show an ammonia concentration of 170 μmol/L (N < 30) and low serum citrulline levels. The oral intake of which of the following nutrients should be restricted in this patient?
A 16-year-old boy presents with acute left-sided weakness. The patient is obtunded and can not provide any history other than his stomach hurts. The patient’s friend states that the patient has had episodes like this in the past and that “he has the same weird disease as his mom”. On physical examination, strength is 1 out of 5 in the left upper and lower extremities. A noncontrast CT scan of the head is normal. Laboratory tests reveal an anion gap metabolic acidosis. Which of the following is a normal function of the structure causing this patient’s condition?
A 40-year-old woman comes to the physician because of a 6-day history of painless blisters on her hands, forearms, and face. Some of the blisters have popped and released a clear fluid. She is otherwise healthy. She had been working the night shift as a security guard for the past few years and switched to the day shift 2 weeks ago. She started wearing a new metal wristwatch last week. Her mother had a similar rash in the past. Her only medication is an estrogen-based oral contraceptive. She drinks 2 beers every night and occasionally more on the weekends. She used intravenous heroin in the past but stopped 20 years ago. Vital signs are within normal limits. Examination shows bullae and oozing erosions in different stages of healing on her arms, dorsal hands, ears, and face. Oral examination shows no abnormalities. There are some atrophic white scars and patches of hyperpigmented skin on the arms and face. Further evaluation of this patient is most likely to show which of the following findings?
A 4-day-old male newborn delivered at 39 weeks' gestation is evaluated because of poor feeding, recurrent vomiting, and lethargy. Physical examination shows tachypnea with subcostal retractions. An enzyme assay performed on a liver biopsy specimen shows decreased activity of carbamoyl phosphate synthetase I. This enzyme plays an important role in the breakdown and excretion of amino groups that result from protein digestion. Which of the following is an immediate substrate for the synthesis of the molecule needed for the excretion of amino groups?
A 2-day-old male is seen in the newborn nursery for repeated emesis and lethargy. He was born at 39 weeks to a 24-year-old mother following an uncomplicated pregnancy and birth. He has been breastfeeding every 2 hours and has 10 wet diapers per day. His father has a history of beta-thalassemia minor. Laboratory results are as follows: Hemoglobin: 12 g/dL Platelet count: 200,000/mm³ Mean corpuscular volume: 95 µm³ Reticulocyte count: 0.5% Leukocyte count: 5,000/mm³ with normal differential Serum: Na+: 134 mEq/L Cl-: 100 mEq/L K+: 3.3 mEq/L HCO3-: 24 mEq/L Urea nitrogen: 1 mg/dL Creatinine: 0.6 mg/dL Ammonia: 150 µmol/L (normal: 50-80 µmol/L) Which of the following is the most likely diagnosis?
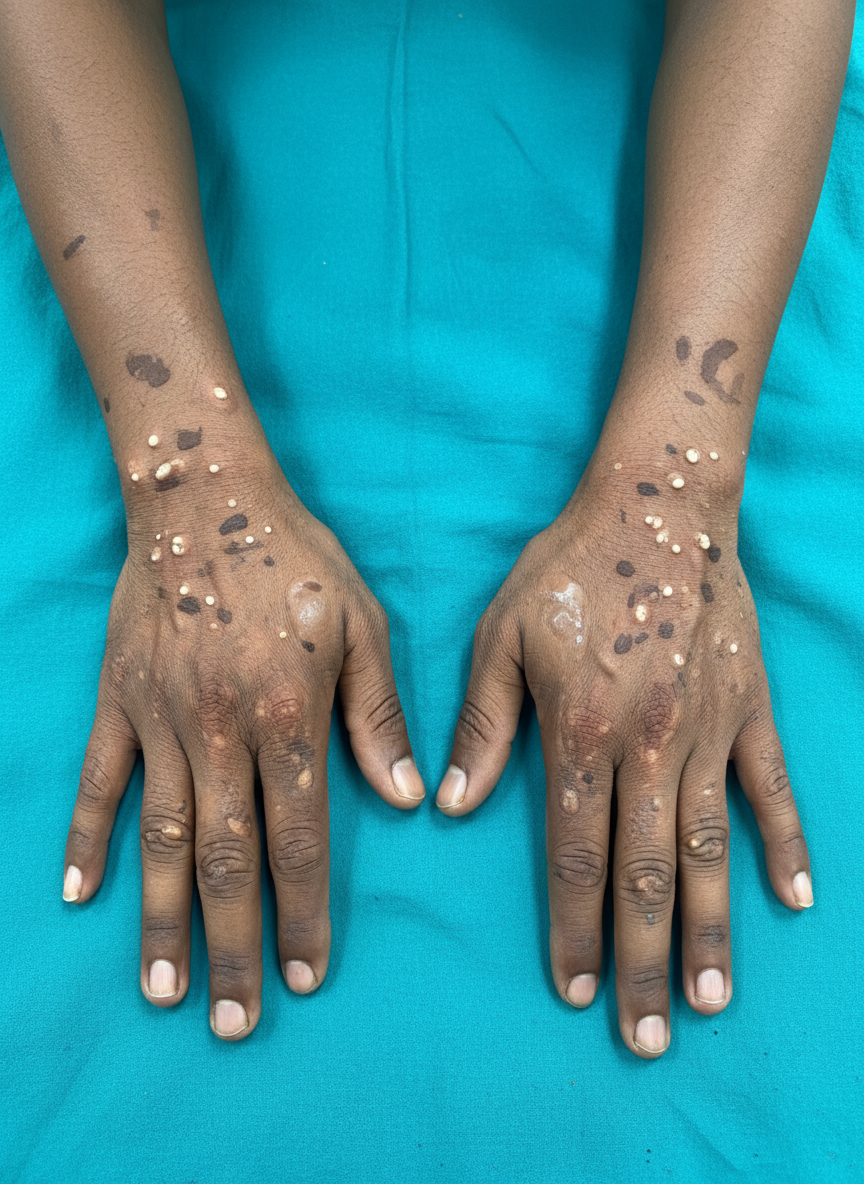
A 51-year-old Indian man visits his physician because of blisters that have appeared on both hands over the past 2 months. The patient states that he works outdoors on freeways and highways, re-paving cracked or otherwise damaged roads. Three months ago, he was working with his crew and felt a sharp pain in his thighs and lower back, which he assumed was caused by the large loads of cement he was carrying to and from his truck. He has been self-medicating with over-the-counter non-steroidal anti-inflammatories, specifically naproxen, twice daily since then. He states that the naproxen relieves his back pain, but he now has blisters on both hands that worry him. On examination, the skin on his face and extremities is healthy and normal-appearing. There are a number of 2-mm-diameter hyperpigmented scars and several bullae overlying normal skin on the dorsal surface of both hands (see image). There are also several small white papules surrounding the hyperpigmented scars. Which of the following is the next step in this patient’s management?
A 20-year-old male presents with confusion, asterixis, and odd behavior. Very early in the morning, his mother found him urinating on the floor of his bedroom. A detailed history taken from the mother revealed that he has been a vegetarian his entire life but decided to "bulk up" by working out and consuming whey protein several times a day. A blood test revealed increased levels of ammonia and orotic acid but a decreased BUN. The patient began hemodialysis and was given oral sodium benzoate and phenylbutyrate, which improved his condition. This patient's condition would be corrected by gene therapy targeting the enzyme that produces which of the following?
Practice by Chapter
Overview and purpose of urea cycle
Practice Questions
Urea cycle reactions and enzymes
Practice Questions
Nitrogen sources and transport forms
Practice Questions
Compartmentalization between mitochondria and cytosol
Practice Questions
Energy requirements of urea cycle
Practice Questions
Regulation of urea cycle
Practice Questions
Integration with TCA cycle
Practice Questions
Hyperammonemia causes and effects
Practice Questions
Urea cycle disorders
Practice Questions
Alternative nitrogen excretion pathways
Practice Questions
Diagnostic markers of urea cycle function
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app