
Signal transduction pathways — MCQs

On this page
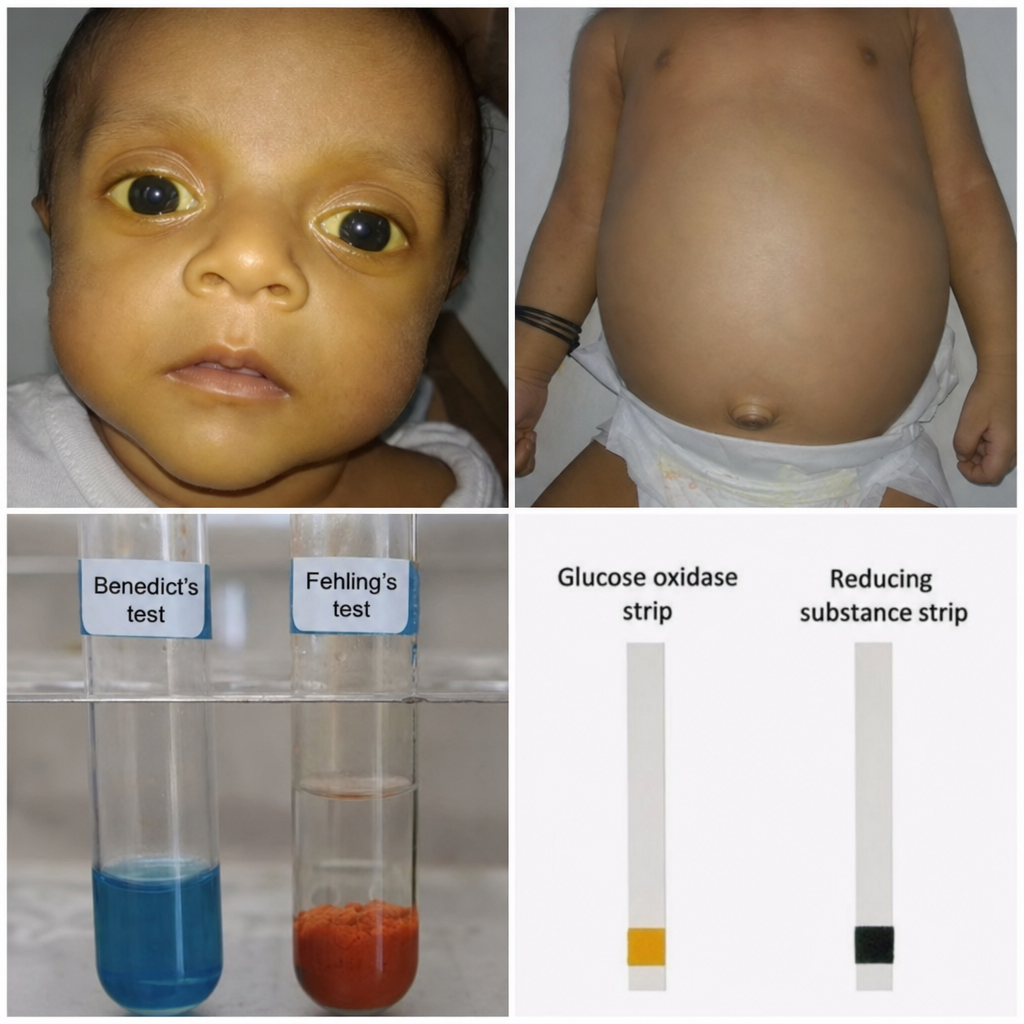
A 6-week-old infant presents with vomiting, jaundice, hepatomegaly, and cataracts. Urine reducing substances are positive, but glucose oxidase strip is negative. The infant is being breastfed. Red blood cell galactose-1-phosphate levels are markedly elevated. Based on the biochemical findings, which enzyme is most likely deficient?
A research team discovers a novel bacterial toxin that causes severe hypotension in infected patients. In vitro studies show the toxin ADP-ribosylates a specific amino acid on Gq alpha subunits, preventing their activation by GPCRs. Patients develop hypotension despite elevated levels of vasopressin, angiotensin II, and endothelin-1. Synthesize the pathophysiological mechanism explaining why multiple vasopressor hormones fail to maintain blood pressure in these patients.
A 42-year-old woman with metastatic melanoma develops severe colitis while being treated with ipilimumab (anti-CTLA-4 antibody) and nivolumab (anti-PD-1 antibody). Her oncologist must decide whether to continue immunotherapy or treat the colitis with immunosuppression. Tumor analysis shows high PD-L1 expression and BRAF wild-type status. Previous conventional chemotherapy failed. Evaluate the optimal management strategy considering signal transduction implications.
A 35-year-old man with familial adenomatous polyposis (FAP) undergoes genetic counseling. He has a germline APC mutation and asks about cancer risk for his children. His physician explains that APC normally regulates beta-catenin levels. Evaluate which downstream transcriptional consequence most directly results from loss of functional APC protein in colonic epithelial cells.
A 58-year-old woman with HER2-positive metastatic breast cancer initially responds to trastuzumab (anti-HER2 antibody) but develops resistance after 18 months. Tumor analysis reveals increased expression of HER3 and continued PI3K/AKT pathway activation despite HER2 blockade. Analyze the mechanism underlying this adaptive resistance.
Practice by Chapter
G-protein coupled receptors
Practice Questions
Tyrosine kinase receptors
Practice Questions
Second messenger systems (cAMP, cGMP, Ca2+)
Practice Questions
MAP kinase pathways
Practice Questions
JAK-STAT pathway
Practice Questions
PI3K-Akt pathway
Practice Questions
Nuclear receptor signaling
Practice Questions
WNT signaling pathway
Practice Questions
Notch signaling pathway
Practice Questions
Hedgehog signaling pathway
Practice Questions
TGF-β/SMAD signaling
Practice Questions
NF-κB signaling
Practice Questions
Dysregulation in disease states
Practice Questions
Pharmacological targeting of signaling pathways
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app