
Molecular Genetics — MCQs

On this page
A 16-year-old boy is brought to the emergency department after losing consciousness. He had no preceding chest pain or palpitations. His father has cataracts and had frontal balding in his twenties but has no history of cardiac disease. His paternal grandfather also had early-onset balding. His pulse is 43/min. Physical examination shows frontal hair loss, temporal muscle wasting, and testicular atrophy. Neurologic examination shows bilateral foot drop and weakness of the intrinsic hand muscles. An ECG shows bradycardia with third-degree atrioventricular block. The severity of this patient's symptoms compared to that of his father is most likely due to which of the following genetic properties?
An 8-year-old African American girl is brought to the clinic by her mother for her regular blood exchange. They come in every 2–3 months for the procedure. The child is in good health with no symptoms. Her last trip to the emergency department was 6 months ago due to bone pain. She was treated with morphine and oxygen and a blood transfusion. She takes hydroxyurea and folic acid daily. She has an uncle that also has to get blood exchanges. Today, her heart rate is 90/min, respiratory rate is 17/min, blood pressure is 110/65 mm Hg, and temperature is 37.0°C (98.6°F). She calmly waits for the machine to be set up and catheters inserted into both of her arms. She watches a movie as her blood is slowly replaced with 6 L of red blood cells. Based on this history, which of the following mechanisms most likely explains this patient's condition?
A 34-year-old man comes to the physician because of blurry vision and fatigue for 2 months. During this period, he has also had occasional bleeding from his gums after brushing his teeth. One month ago, he was diagnosed with deep vein thrombosis after returning from an overseas business meeting. His pulse is 118/min, respirations are 19/min, and blood pressure is 149/91 mm Hg. Pulse oximetry on room air shows an oxygen saturation of 97%. Examination shows bluish discoloration of the lips. The tip of the spleen is palpable 1 cm below the left costal margin. Sensory examination of the hands shows paresthesia. Hemoglobin concentration is 18 g/dL, hematocrit is 65%, leukocytes are 15,000/μL, and platelets are 470,000/μL. His serum erythropoietin concentration is decreased. Activation of which of the following is the most likely underlying cause of this patient's condition?
A 5-year-old boy is brought to the emergency department after he fell on the playground in kindergarten and was unable to get up. His right leg was found to be bent abnormally at the femur, and he was splinted on site by first responders. His past medical history is significant for multiple prior fractures in his left humerus and femur. Otherwise, he has been hitting normal developmental milestones and appears to be excelling in kindergarten. Physical exam also reveals the finding shown in figure A. Which of the following is the most likely cause of this patient's multiple fractures?
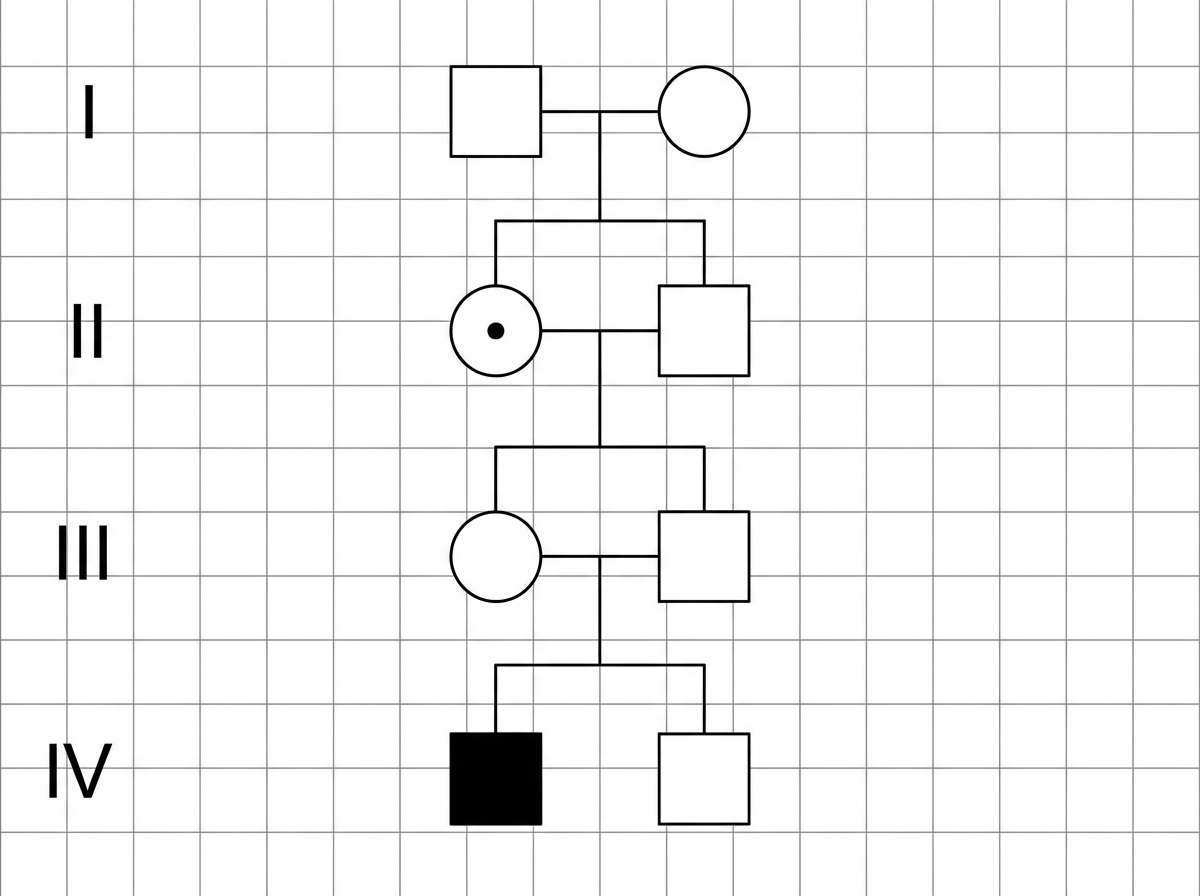
A 28-year-old woman, gravida 1, para 0, at 20 weeks' gestation comes to the physician for genetic counseling. Her brother and maternal uncle both have anemia that worsens after taking certain medications. Based on the pedigree shown, what is the probability that her son will be affected by the disease?
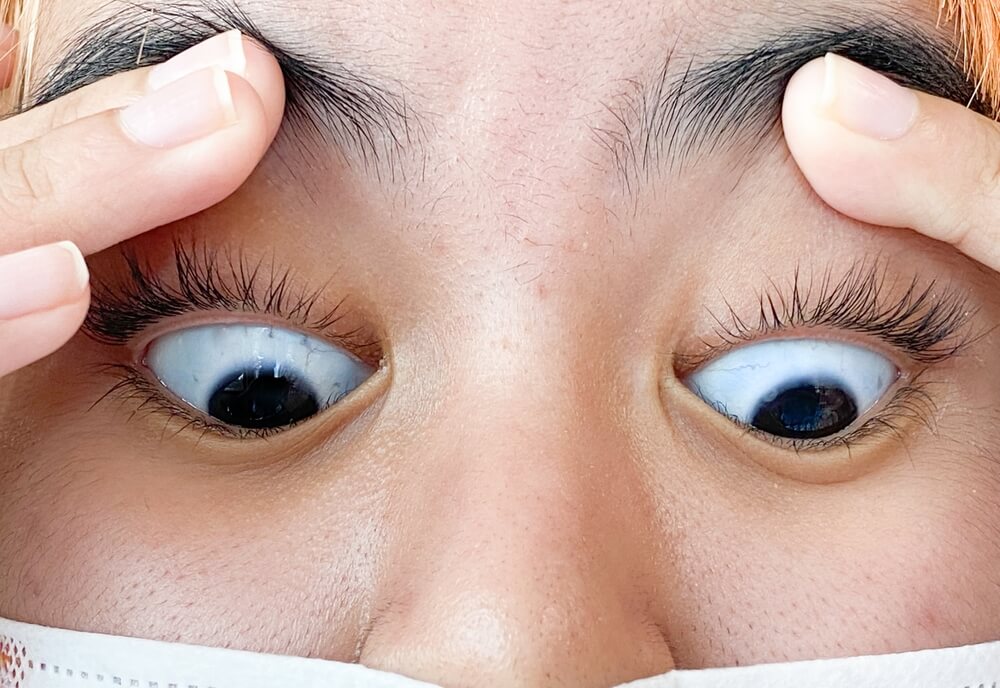
A 10-month-old boy is brought to the physician by his mother for evaluation of abnormal growth and skin abnormalities. His mother has also noticed that his eyes do not fully close when sleeping. He is at the 24th percentile for height, 17th percentile for weight, and 29th percentile for head circumference. Physical examination shows wrinkled skin, prominent veins on the scalp and extremities, and circumoral cyanosis. Genetic testing shows a point mutation in a gene that encodes for a scaffold protein of the inner nuclear membrane. The mutation causes a deformed and unstable nuclear membrane, which leads to premature aging. Which of the following is most likely to be the defective protein?
An 8-year old boy is brought into clinic for evaluation of possible scoliosis that was newly found on a routine exam at school. On exam, he is also noted to be in the 99th percentile for height and 70th percentile for weight. He appears to have abnormally long extremities as well as an upward lens dislocation on ophthalmologic exam. A mutation leading to a defect in which of the following proteins is the most likely cause of his condition?
A 48-year-old man presents to a physician with complaints of paresthesia of the lower extremities, which he has had for the last 3 months. He has been frequently fatigued for the past 5 months and also experienced an increased frequency of urination over the last few months. There is no history of a known medical condition or of substance abuse. His physical examination does not reveal any specific abnormality, except that he is obese: his body mass index is 34.6 kg/m2. The patient’s detailed laboratory evaluation reveals a fasting plasma glucose of 160 mg/dL and 2-hour plasma glucose of 270 mg/dL. His physician tells him that his laboratory evaluation suggests a diagnosis of diabetes mellitus type 2. The patient, surprised by this news, asks his physician why he has developed diabetes mellitus even though no one else in his family has ever suffered from it. The physician explains to him that genetic factors play an important role in the development of diabetes mellitus, but that their interactions are complex. Apart from neonatal diabetes mellitus and maturity-onset diabetes of the young (MODY), the development of diabetes mellitus cannot be explained by a single genetic mutation. Which of the following options best explains the genetics of the form of diabetes mellitus from which this man is suffering?
A 33-year-old man presents to his physician with a 3-year history of gradually worsening tics and difficulty walking. He was last seen by the physician 5 years ago for anxiety, and he has been buying anti-anxiety medications from an internet website without a prescription as he cannot afford to pay for doctor’s visits. Now, the patient notes that his anxiety is somewhat controlled, but motor difficulties are making it difficult for him to work and socialize. Family history is unobtainable as his parents died in an accident when he was an infant. He grew up in foster care and was always a bright child. An MRI of the brain is ordered; it shows prominent atrophy of the caudate nucleus. Repeats of which of the following trinucleotides are most likely responsible for this patient’s disorder?
An 18-month-old boy is brought in by his parents for a routine check-up. The parents state that the patient still has not had any language development, and they are concerned about developmental delay. Of note, they have also noticed that the patient’s facial features have changed significantly in the last year. The patient also seems to have trouble visually focusing on objects or on the television. On exam, the patient's temperature is 98.2°F (36.8°C), blood pressure is 108/72 mmHg, pulse is 86/min, and respirations are 14/min. Of interest, the patient has not increased much in length or weight in the past 3 months. He is now in the 25th percentile for weight but is in the 90th percentile for head circumference. The patient does not appear to have any gross or fine motor deficiencies. Of note, he has coarse facial features that were not previously noted, including a long face, prominent forehead, and protruding eyes. The patient has corneal clouding bilaterally. At rest, the patient keeps his mouth hanging open. After extensive workup, the patient is found to have 2 mutated copies of the IDUA gene, with no production of the protein iduronidase. Which of the following is the likely mutation found in this disease?
Practice by Chapter
DNA structure and organization
Practice Questions
Chromosomal structure
Practice Questions
DNA replication
Practice Questions
Cell cycle and mitosis
Practice Questions
Meiosis and genetic recombination
Practice Questions
Mutation types and consequences
Practice Questions
Mendelian inheritance patterns
Practice Questions
Non-Mendelian inheritance
Practice Questions
Genetic linkage and mapping
Practice Questions
Population genetics principles
Practice Questions
Genetic polymorphisms
Practice Questions
Gene therapy approaches
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app