
Molecular Genetics — MCQs

On this page
A 14-year-old Caucasian girl presents to the pediatrician for poor balance. She reports a 7-month history of frequent falls that has progressively worsened. She has fallen 3 times in the past week and feels like she cannot walk normally. She was born full-term and spent 2 days in the neonatal intensive care unit for respiratory distress. She has had an otherwise normal childhood. Her family history is notable for multiple cardiac deaths before the age of 60. Her mother had a posterior spinal fusion for kyphoscoliosis as an adolescent. On exam, the patient has 4/5 strength in her bilateral upper and lower extremities. She walks with a staggering gait. Pes cavus is appreciated bilaterally. Skin examination is normal. This patient has a condition that is caused by a trinucleotide repeat of which of the following nucleotides?
A team of biology graduate students are performing research on epigenetics and chromosome inactivation. The goal is to silence all the genes on a chromosome at once. The team chooses to develop a model based on a known human gene that can accomplish this task in vivo. Which of the genes listed below would be a suitable model for their research?
A 4-year-old girl is brought to the emergency department after falling off a chair and injuring her right leg. During the past 2 years, she has had two long bone fractures. She is at the 5th percentile for height and 20th percentile for weight. Her right lower leg is diffusely erythematous. The patient withdraws and yells when her lower leg is touched. A photograph of her face is shown. An x-ray of the right lower leg shows a transverse mid-tibial fracture with diffusely decreased bone density. Which of the following is the most likely cause of this patient's symptoms?
An investigator is studying the effect of chromatin structure on gene regulation. The investigator isolates a class of proteins that compact DNA by serving as spools upon which DNA winds around. These proteins are most likely rich in which of the following compounds?
A 1-year-old boy brought in by his mother presents to his physician for a routine checkup. On examination, the child is happy and playful and meets normal cognitive development markers. However, the child’s arms and legs are not meeting development goals, while his head and torso are. The mother states that the boy gets this from his father. Which of the following is the mutation associated with this presentation?
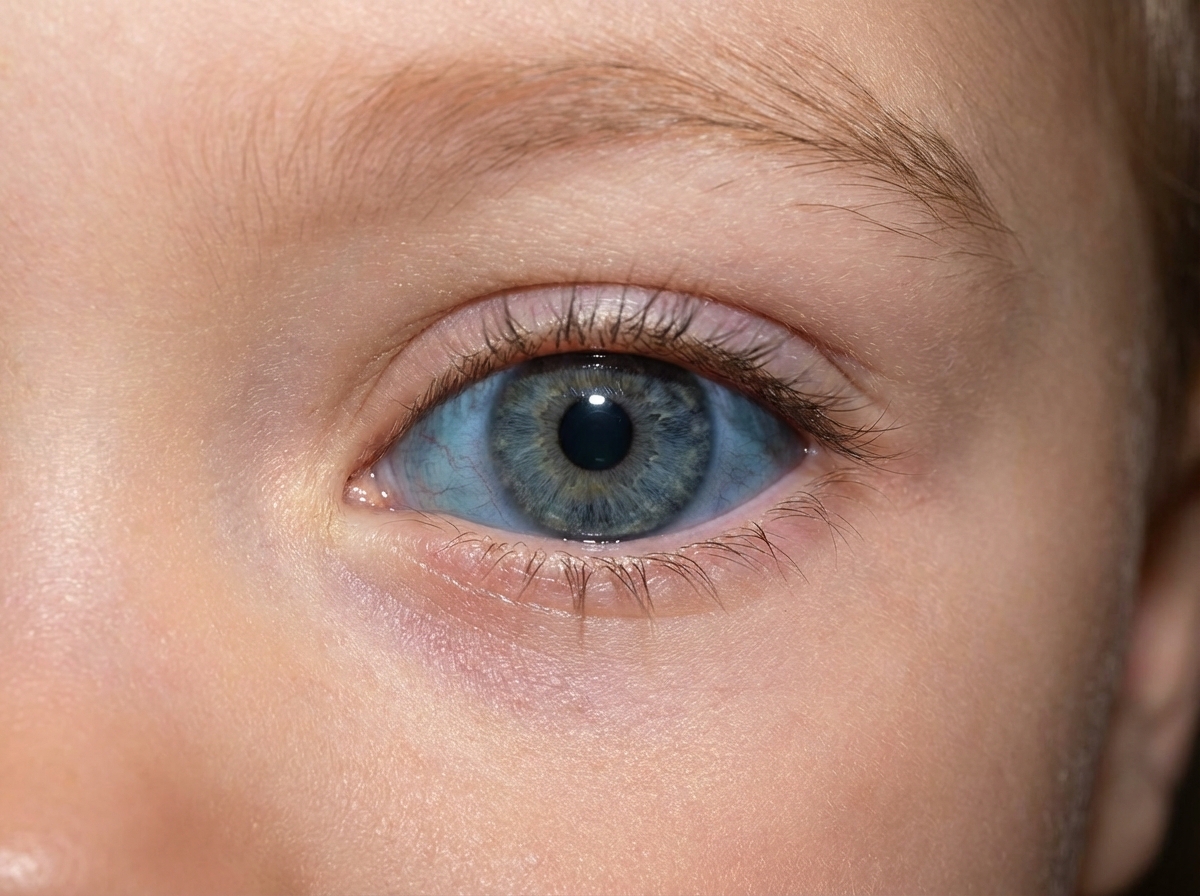
An 11-year-old male with light purple eyes presents with gradual loss of bilateral visual acuity. Over the past several years, vision has worsened from 20/20 to 20/100 in both eyes. He also has mild nystagmus when focusing on objects such as when he is trying to do his homework. He is diagnosed with a disease affecting melanin production in the iris. If both of his parents are unaffected, which of the following represents the most likely probabilities that another male or female child from this family would be affected by this disorder?
A baby is delivered at 39 weeks without complications. Upon delivery, there are obvious craniofacial abnormalities, including micrognathia, cleft lip, and cleft palate. On further inspection, downward slanting eyes and malformed ears are seen. The child has an APGAR score of 9 and 9 at 1 and 5 minutes respectively. There are no signs of cyanosis or evidence of a heart murmur. Which of the following is the most likely underlying cause of this patient’s presentation at birth?
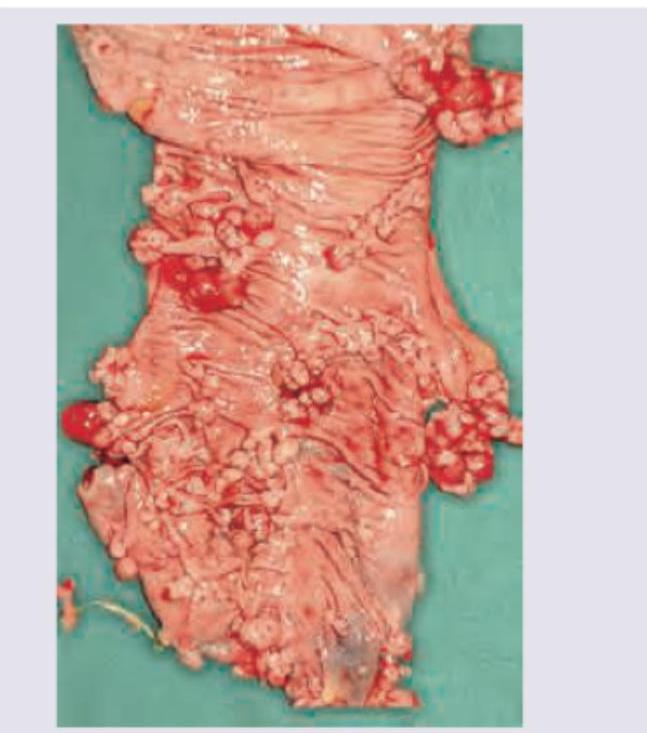
A previously healthy 42-year-old man comes to the emergency room with constipation and diffuse, worsening abdominal pain for 2 days. He has no history of major medical illness. His father died in a car accident at the age of 32 years, and his mother has type 2 diabetes mellitus. A diagnosis of bowel obstruction is suspected and he is taken to the operating room for exploratory laparotomy. A partial resection of the colon is performed. The gross appearance of the patient's colonic tissue is shown. Microscopic examination shows tubular, tubulovillous, and villous adenomas. Assuming the patient's partner is not a carrier of the condition, which of the following is the likelihood that this patient’s children will develop this condition?
Researchers are investigating oncogenes, specifically the KRAS gene that is associated with colon, lung, and pancreatic cancer. They have established that the gain-of-function mutation in this gene increases the chance of cancer development. They are also working to advance the research further to study tumor suppressor genes. Which of the genes below is considered a tumor suppressor gene?
An investigator is studying the incidence of sickle cell trait in African American infants. To identify the trait, polymerase chain reaction testing is performed on venous blood samples obtained from the infants. Which of the following is required for this laboratory technique?
Practice by Chapter
DNA structure and organization
Practice Questions
Chromosomal structure
Practice Questions
DNA replication
Practice Questions
Cell cycle and mitosis
Practice Questions
Meiosis and genetic recombination
Practice Questions
Mutation types and consequences
Practice Questions
Mendelian inheritance patterns
Practice Questions
Non-Mendelian inheritance
Practice Questions
Genetic linkage and mapping
Practice Questions
Population genetics principles
Practice Questions
Genetic polymorphisms
Practice Questions
Gene therapy approaches
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app