
Molecular Genetics — MCQs

On this page
A 4-year-old child presents with developmental delay, ataxia, and inappropriate laughter. The parents undergo genetic testing to determine the cause of their child's symptoms. Results show no mutations in all three family members that would cause this constellation of symptoms. Karyotyping reveals no deletions, insertions, or gene translocations. However, methylation studies demonstrate abnormal imprinting patterns at the 15q11-q13 region. Based on these findings, the child is diagnosed with Angelman syndrome. Which of the following genetic mechanisms best describes the cause of this disorder?
A 20-year-old female presents to the emergency department with squeezing right upper quadrant pain worse after eating. She has a history of a microcytic, hypochromic anemia with target cells. Physical exam shows severe tenderness to palpation in the right upper quadrant and a positive Murphy's sign. By genetic analysis a single point mutation is detected in the gene of interest. Despite this seemingly minor mutation, the protein encoded by this gene is found to be missing a group of 5 consecutive amino acids though the amino acids on either side of this sequence are preserved. This point mutation is most likely located in which of the following regions of the affected gene?
A 2-year-old boy from a rural community is brought to the pediatrician after his parents noticed a white reflection in both of his eyes in recent pictures. Physical examination reveals bilateral leukocoria, nystagmus, and inflammation. When asked about family history of malignancy, the father of the child reports losing a brother to an eye tumor when they were children. With this in mind, which of the following processes are affected in this patient?
A 38-year-old woman seeks evaluation at the emergency room for sudden onset of pain and swelling of her left leg since last night. Her family history is significant for maternal breast cancer (diagnosed at 52 years of age) and a grandfather with bronchioloalveolar carcinoma of the lungs at 45 years of age. When the patient was 13 years old, she was diagnosed with osteosarcoma of the right distal femur that was successfully treated with surgery. The physical examination shows unilateral left leg edema and erythema that was tender to touch and warm. Homan's sign is positive. During the abdominal examination, you also notice a large mass in the left lower quadrant that is firm and fixed with irregular borders. Proximal leg ultrasonography reveals a non-compressible femoral vein and the presence of a thrombus after color flow Doppler evaluation. Concerned about the association between the palpable mass and a thrombotic event in this patient, you order an abdominal CT scan with contrast that reports a large left abdominopelvic cystic mass with thick septae consistent with ovarian cancer, multiple lymph node involvement, and ascites. Which of the following genes is most likely mutated in this patient?
A 16-year-old boy presents with a long-standing history of anemia. Past medical history is significant for prolonged neonatal jaundice and multiple episodes of jaundice without fever. On physical examination, the patient shows generalized pallor, scleral icterus, and splenomegaly. His hemoglobin is 10 g/dL, and examination of a peripheral blood smear shows red cell basophilic stippling. Which of the following is the most likely diagnosis in this patient?
A 24-year-old man is referred to an endocrinologist for paroxysms of headaches associated with elevated blood pressure and palpitations. He is otherwise healthy, although he notes a family history of thyroid cancer. His physical examination is significant for the findings shown in Figures A, B, and C. His thyroid is normal in size, but there is a 2.5 cm nodule palpable in the right lobe. On further workup, it is found that he has elevated plasma-free metanephrines and a normal TSH. Fine-needle aspiration of the thyroid nodule stains positive for calcitonin. The endocrinologist suspects a genetic syndrome. What is the most likely inheritance pattern?
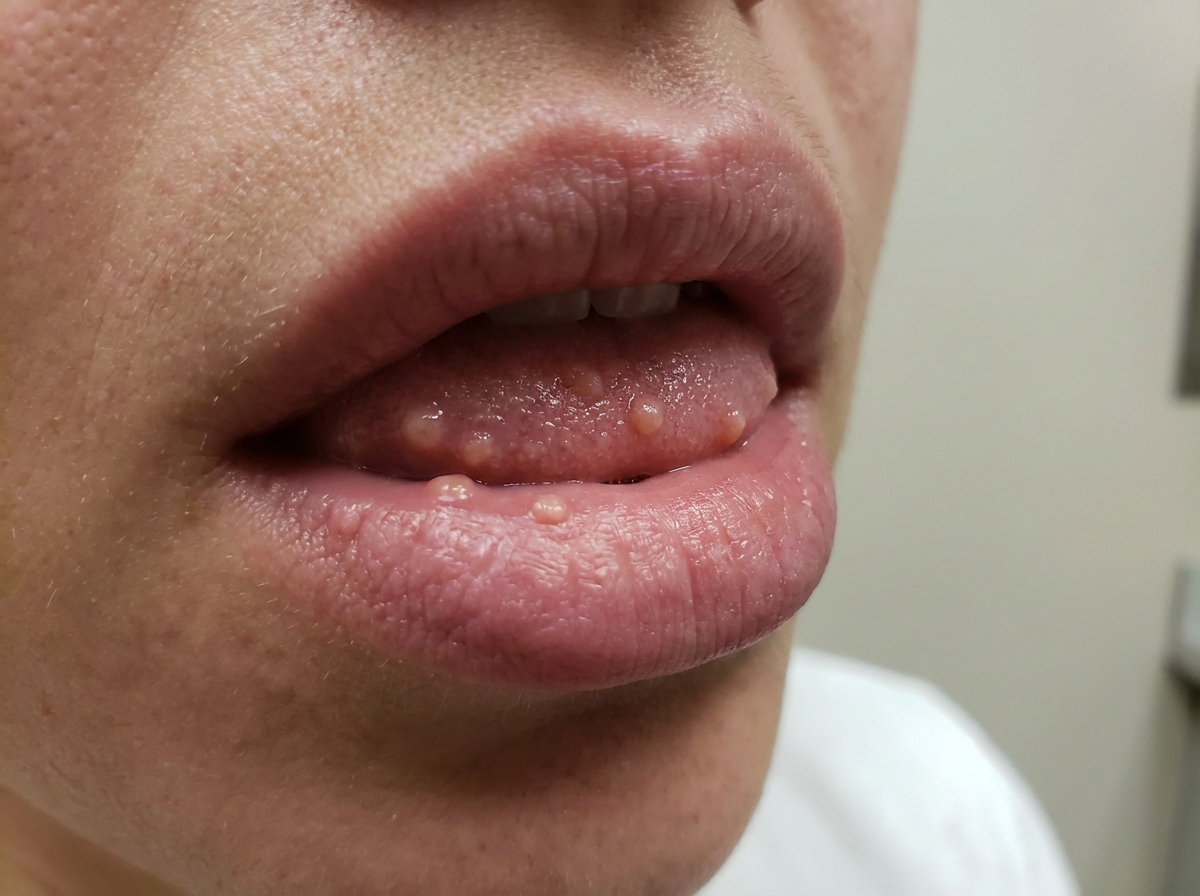
A 3-year-old boy is brought to the physician by his parents for the evaluation of easy bruising for several months. Minor trauma also causes scratches that bleed. Two months ago, a fall from his bed caused a large forehead hematoma and a left elbow laceration. He sometimes does not eat because of pain while chewing. Vital signs are within normal limits. Examination shows that the skin can be stretched further than normal and is fragile. Range of motion of the joints is slightly increased. There is tenderness to palpation of the temporomandibular joints bilaterally. Which of the following is the most likely underlying cause of this patient's symptoms?
An investigator is studying the structure of the amino-terminal of the Huntingtin protein using x-ray crystallography. The terminal region is determined to have an α-helix conformation. Which of the following forces is most likely responsible for maintaining this conformation?
A 23-year-old man presents to his primary care physician with 2 weeks of headache, palpitations, and excessive sweating. He has no past medical history and his family history is significant for clear cell renal cell carcinoma in his father as well as retinal hemangioblastomas in his older sister. On presentation his temperature is 99°F (37.2°C), blood pressure is 181/124 mmHg, pulse is 105/min, and respirations are 18/min. After administration of appropriate medications, he is taken emergently for surgical removal of a mass that was detected by abdominal computed tomography scan. A mutation on which of the following chromosomes would most likely be seen in this patient?
A 13-year-old female presents to the emergency room complaining of severe abdominal pain. She reports acute onset of diffuse abdominal pain twelve hours prior to presentation. She has vomited twice and has not had a bowel movement in that time. She is in the fetal position because it relieves the pain. Her past medical history is notable for asthma; however, she was adopted as a baby and her family history is unknown. Her temperature is 99.7°F (37.6°C), blood pressure is 130/85 mmHg, pulse is 110/min, and respirations are 22/min. Physical examination reveals abdominal distension and tenderness to palpation. A sausage-shaped abdominal mass is palpated in the right upper quadrant. Mucocutaneous blue-gray macules are evident on the child’s buccal mucosa. A mutation in which of the following genes is associated with this patient’s condition?
Practice by Chapter
DNA structure and organization
Practice Questions
Chromosomal structure
Practice Questions
DNA replication
Practice Questions
Cell cycle and mitosis
Practice Questions
Meiosis and genetic recombination
Practice Questions
Mutation types and consequences
Practice Questions
Mendelian inheritance patterns
Practice Questions
Non-Mendelian inheritance
Practice Questions
Genetic linkage and mapping
Practice Questions
Population genetics principles
Practice Questions
Genetic polymorphisms
Practice Questions
Gene therapy approaches
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app