
Molecular Genetics — MCQs

On this page
A Caucasian 32-year-old woman has an uncomplicated vaginal delivery, giving birth to male and female fraternal twins at term. At 2 days of life, the twin sister develops abdominal distension without emesis, and the mother states that she has not noticed the passage of stool for this infant. Genetic testing identifies deletion of an amino acid in a membrane channel for the girl. Both parents are healthy. Assuming that twin brother's disease status/symptomatology is unclear, which of the following best approximates the probability that the twin brother is a carrier of the disease allele?
A healthy 30-year-old woman comes to the physician with her husband for preconception counseling. Her husband is healthy but she is concerned because her brother was recently diagnosed with a genetic liver condition for which he takes penicillamine. Her father-in-law has liver cirrhosis and a tremor. The results of genetic testing show that both the patient and her husband are carriers of a mutation in the ATP7B gene. Which of the following is the chance that this patient’s offspring will eventually develop the hereditary condition?
A 2-month-old boy is presented to the clinic for a well-child visit by his parents. They are concerned with his weak cry and difficulty with feeding. Birth history reveals that the boy was born at the 37th week of gestation by cesarean section due to poor fetal movement and fetal distress. His Apgar scores were 3 and 5 at 1st and 5th minute respectively and his birth weight was 2.5 kg (6 lb). His vital signs include heart rate 120/min, respiratory rate 40/min, blood pressure 90/50 mm Hg, and temperature 37.0°C (98.6°F). Physical examination reveals a malnourished boy with a small narrow forehead and a small jaw. His mouth is small and he has comparatively small genitals. He has a poor muscle tone. After repeated follow-up, he gains weight rapidly but his height fails to increase. Developmental milestones are delayed at the age of 3 years. Genetic testing reveals Prader-Willi syndrome. Which of the following is the most common mechanism for the development of this patient’s condition?
An investigator studying DNA replication in Campylobacter jejuni inoculates a strain of this organism into a growth medium that contains radiolabeled thymine. After 2 hours, the rate of incorporation of radiolabeled thymine is measured as a proxy for the rate of DNA replication. The cells are then collected by centrifugation and suspended in a new growth medium that lacks ribonucleotides. After another 2 hours, the rate of incorporation of radiolabeled thymine is measured again. The new growth medium directly affects the function of which of the following enzymes?
A 32-year-old man presents to his primary care provider reporting weakness. He recently noticed that he has difficulty letting go of a doorknob or releasing his hand after shaking hands with others. His past medical history is notable for diabetes, for which he takes metformin. He drinks 2-3 beers per day, uses marijuana occasionally, and works as a security guard. His family history is notable for an early cardiac death in his father. His temperature is 98.6°F (37°C), blood pressure is 130/85 mmHg, pulse is 85/min, and respirations are 18/min. On exam, there is notable muscle atrophy in his hands, feet, and neck. He has delayed hand grip release bilaterally and is slow to return from a smile to a neutral facial expression. His gait is normal, and Romberg's test is negative. He also has frontal balding. This patient’s condition is caused by a mutation in which of the following genes?
A 13-year-old girl is brought to the outpatient clinic by her parents with a complaint of episodic spasm in her fingers for the past few months. Upon further questioning, her mother notes that the girl has not been doing well at school. She also believes that the girl is shorter than the other children in her class. On examination, her pulse is 72/min, temperature 37.6°C (99.7°F), respiratory rate 16/min, and blood pressure 120/88 mm Hg. The girl has short 4th and 5th fingers on both hands, a round face, and discolored teeth. Her height is 135 cm (4 ft 5 in) and she weighs 60 kg (132 lb). Investigation reports show the following values: Hemoglobin (Hb%) 12.5 g/dL White blood cell total count 10,000/mm3 Platelets 260,000/mm3 Calcium, serum (Ca2+) 4.0 mg/dL Serum albumin 4.0 g/dL Alanine aminotransferase (ALT), serum 15 U/L Aspartate aminotransferase (AST), serum 8 U/L Serum creatinine 0.5 mg/dL Urea 27 mg/dL Sodium 137 mEq/L Potassium 4.5 mEq/L Magnesium 2.5 mEq/L Parathyroid hormone, serum, N-terminal 930 pg/mL (normal: 230-630 pg/mL) Serum vitamin D 45 ng/dL Which of the following is the mode of inheritance of the disease this patient has?
A 1-year-old girl born to a 40-year-old woman is undergoing an examination by a pediatric resident in the hospital. The pregnancy was uneventful and there were no complications during the delivery. The physical examination reveals midface hypoplasia with a flat nasal bridge and upslanting palpebral fissures. She has a small mouth and chest auscultation reveals a blowing holosystolic murmur that is heard best along the sternal border. The family history is unremarkable. A karyotype analysis is ordered because the resident suspects a numerical chromosomal disorder. Which of the following phenomena leads to the infant’s condition?
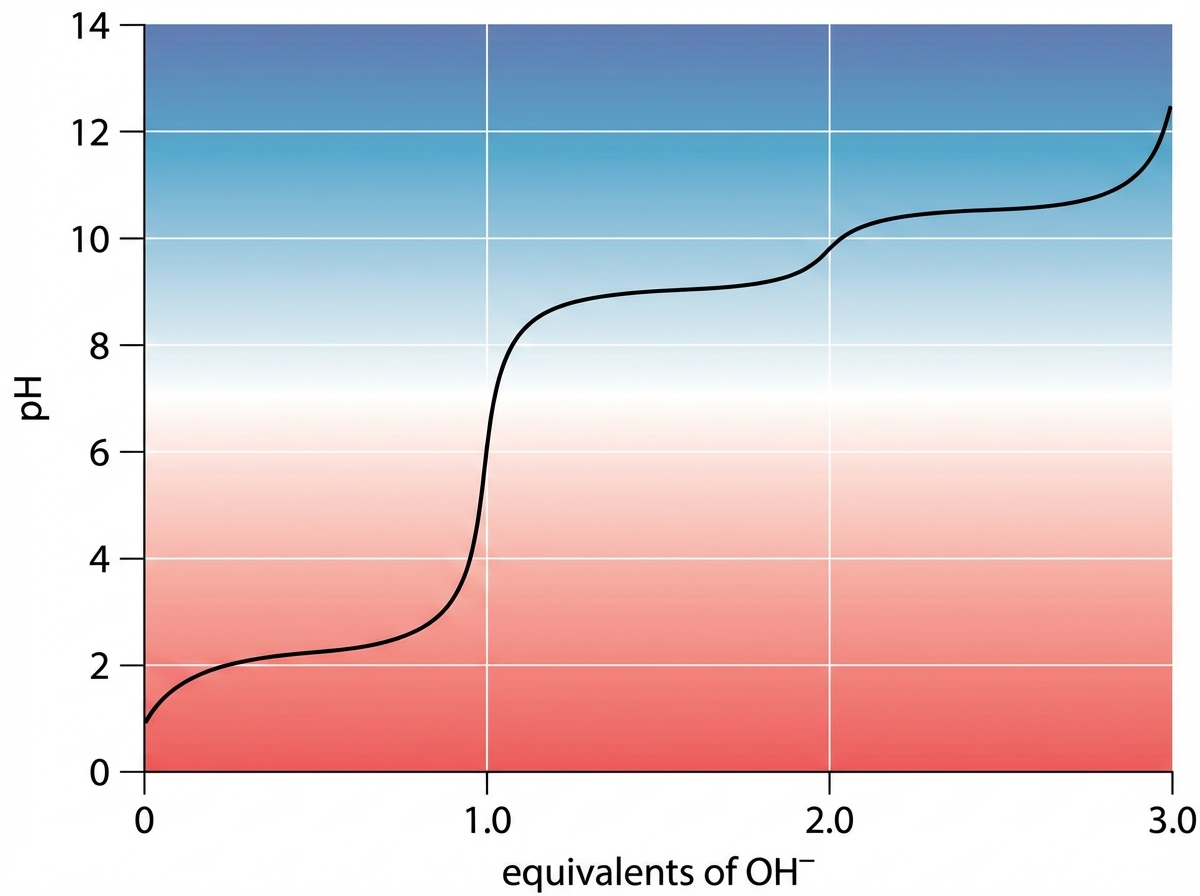
An investigator studying epigenetic mechanisms isolates histone proteins, the structural motifs involved in DNA binding and regulation of transcription. The peptide bonds of histone proteins are hydrolyzed and one type of amino acid is isolated. At normal body pH, this amino acid has a net charge of +1 . The investigator performs titration of this amino acid and obtains the graph shown. The isolated amino acid is most likely which of the following?
A 5-year-old boy presents to the pediatrician after his parents noted that he could not sustain physical exertion and would experience muscle cramping. It was noted that after physical exertion the boy experienced severe muscle pain. After a series of biochemical and genetic tests, it was discovered that the boy had a nonsense mutation in the gene encoding the muscle glycogen phosphorylase. Thus he was diagnosed with McArdle's disease. Which of the following mRNA changes would be expected to cause this mutation?
An 8-year-old boy is brought to the physician for evaluation of developmental delay and recurrent tonic-clonic seizures. There is no family history of seizures or other serious illness. Current medications include risperidone for hyperactivity. He is at the 17th percentile for head circumference. Examination shows protrusion of the mandible, strabismus, and a laughing facial expression. His gait is unsteady. He has a vocabulary of about 200 words and cannot speak in full sentences. Karyotype analysis shows a 46, XY karyotype without chromosomal deletions. Which of the following genetic mechanisms best explains this patient's findings?
Practice by Chapter
DNA structure and organization
Practice Questions
Chromosomal structure
Practice Questions
DNA replication
Practice Questions
Cell cycle and mitosis
Practice Questions
Meiosis and genetic recombination
Practice Questions
Mutation types and consequences
Practice Questions
Mendelian inheritance patterns
Practice Questions
Non-Mendelian inheritance
Practice Questions
Genetic linkage and mapping
Practice Questions
Population genetics principles
Practice Questions
Genetic polymorphisms
Practice Questions
Gene therapy approaches
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app