
Molecular Genetics — MCQs

On this page
A 10-year-old boy is brought to the physician because of recurring episodes of achy muscle pain in his legs. He has a history of poor school performance despite tutoring and has been held back two grades. He is at the 40th percentile for height and 30th percentile for weight. Examination shows ptosis, a high-arched palate, and muscle weakness in the face and hands; muscle strength of the quadriceps and hamstrings is normal. Sensation is intact. Percussion of the thenar eminence causes the thumb to abduct and then relax slowly. Which of the following is the most likely underlying cause?
A 5-year-old boy is brought to the clinic by his mother for an annual check-up. The family recently moved from Nebraska and is hoping to establish care. The patient is home schooled and mom is concerned about her son’s development. He is only able to say 2 to 3 word sentences and has been “behind on his alphabet." He always seems to be disinterested and "just seems to be behind.” The patient is observed to be focused on playing with his cars during the interview. Physical examination demonstrate a well-nourished child with poor eye contact, a prominent jaw, a single palmar crease, and bilaterally enlarged testicles. What is the most likely mechanism of this patient’s findings?
Two healthy adults have only one child. He has Friedreich ataxia (FA). They are considering having more children, but are uncertain of their risk of having another child with the condition. What should they do?
A 64-year-old woman presents to an urgent care clinic with edema of her lips and difficulty breathing. She reports that she had multiple root canals performed earlier today, and she started to notice swelling of her lips 2 hours ago. The symptoms have now progressed to where she is having trouble breathing. She notes similar episodes in the past after minor procedures such as this. The blood pressure is 118/76 mm Hg, the heart rate is 84/min, and the respiratory rate is 16/min. Physical examination is remarkable for edema of her lips and mild inspiratory stridor. The laboratory results are remarkable for a low level of C1 esterase inhibitor. Of the following options, which is the most likely diagnosis?
A 5-year-old boy is brought to the emergency room by his parents after slipping on a rug at home and experiencing exquisite pain and swelling of his arms. Radiographs reveal a new supracondylar fracture of the humerus, as well as indications of multiple, old fractures that have healed. His parents note that an inherited disorder is present in their family history. A comprehensive physical exam also reveals blue-tinted sclera and yellow-brown, discolored teeth. What is the etiology of the patient’s disorder?
A 5-year-old boy is brought to the physician because of behavioral problems. His mother says that he has frequent angry outbursts and gets into fights with his classmates. He constantly complains of feeling hungry, even after eating a full meal. He has no siblings, and both of his parents are healthy. He is at the 25th percentile for height and is above the 95th percentile for weight. Physical examination shows central obesity, undescended testes, almond-shaped eyes, and a thin upper lip. Which of the following genetic changes is most likely associated with this patient's condition?
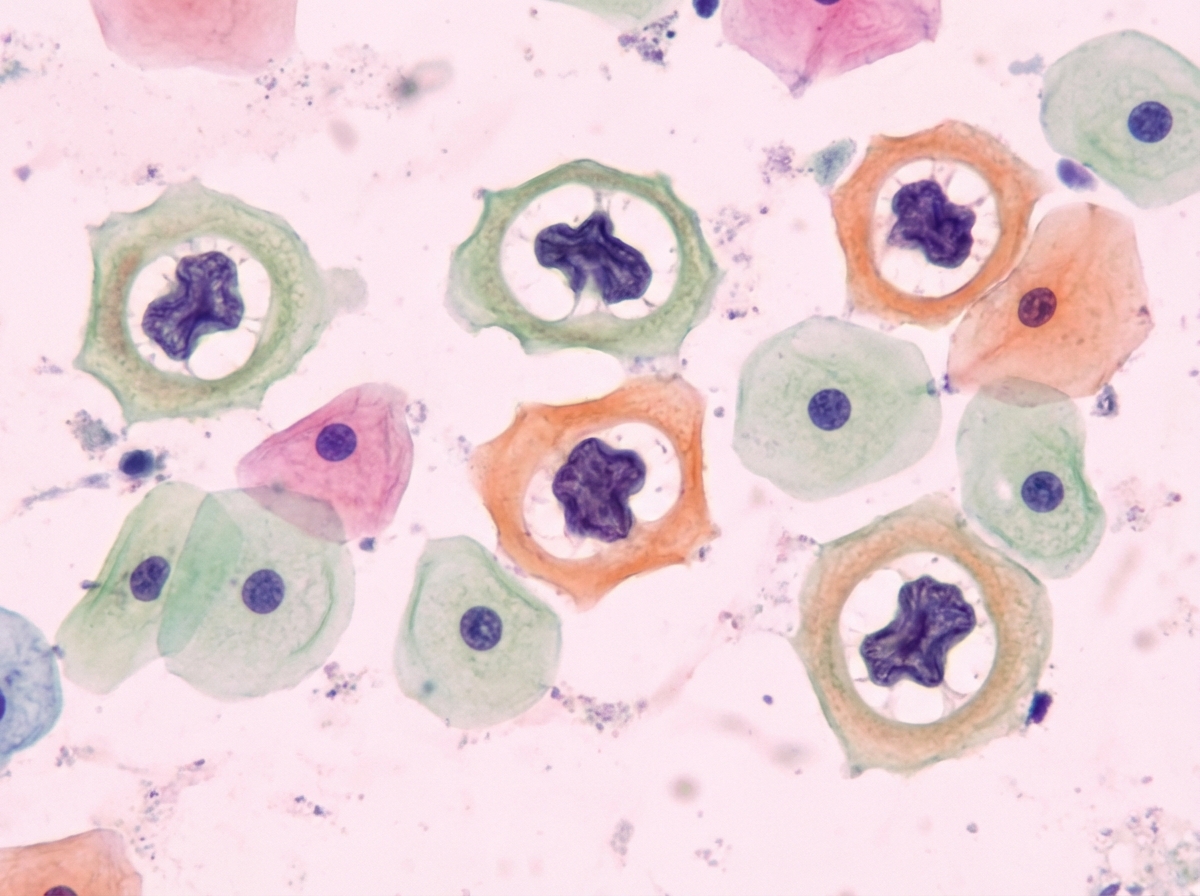
A 29-year-old woman presents to her gynecologist for a routine check-up. She is sexually active with multiple partners and intermittently uses condoms for contraception. She denies vaginal discharge, burning, itching, or rashes in her inguinal region. Pelvic examination is normal. Results from a routine pap smear are shown. The cellular changes seen are attributable to which of the following factors?
A researcher is investigating compounds that modulate the cell cycle as possible chemotherapeutic agents against peripheral T-cell lymphoma. The researcher discovers a group of natural compounds with inhibitory activity against histone deacetylases, a class of enzymes that remove acetyl groups from the lysine residues of histones. A histone deacetylase inhibitor most likely causes which of the following?
A genetic counselor sees a family for the first time for genetic assessment. The 24-year-old businessman and his 19-year-old sister are concerned about having a mutant allele and have decided to get tested. Their grandfather and great aunt both have Huntington’s disease which became apparent when they turned 52. Their father who is 47 years old appears healthy. The geneticist discusses both the benefits and risks of getting tested and orders some tests. Which of the following tests would best provide evidence for whether the siblings are carriers or not?
A mother brings her 3-year-old daughter to the pediatrician because she is concerned about her development. She states that her daughter seemed to regress in her motor development. Furthermore, she states she has been having brief episodes of uncontrollable shaking, which has been very distressing to the family. During the subsequent work-up, a muscle biopsy is obtained which demonstrates red ragged fibers and a presumptive diagnosis of a genetic disease is made. The mother states that she has another 6-year-old son who does not seem to be affected or have any similar symptoms. What genetic term explains this phenomenon?
Practice by Chapter
DNA structure and organization
Practice Questions
Chromosomal structure
Practice Questions
DNA replication
Practice Questions
Cell cycle and mitosis
Practice Questions
Meiosis and genetic recombination
Practice Questions
Mutation types and consequences
Practice Questions
Mendelian inheritance patterns
Practice Questions
Non-Mendelian inheritance
Practice Questions
Genetic linkage and mapping
Practice Questions
Population genetics principles
Practice Questions
Genetic polymorphisms
Practice Questions
Gene therapy approaches
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app