
Molecular Genetics — MCQs

On this page
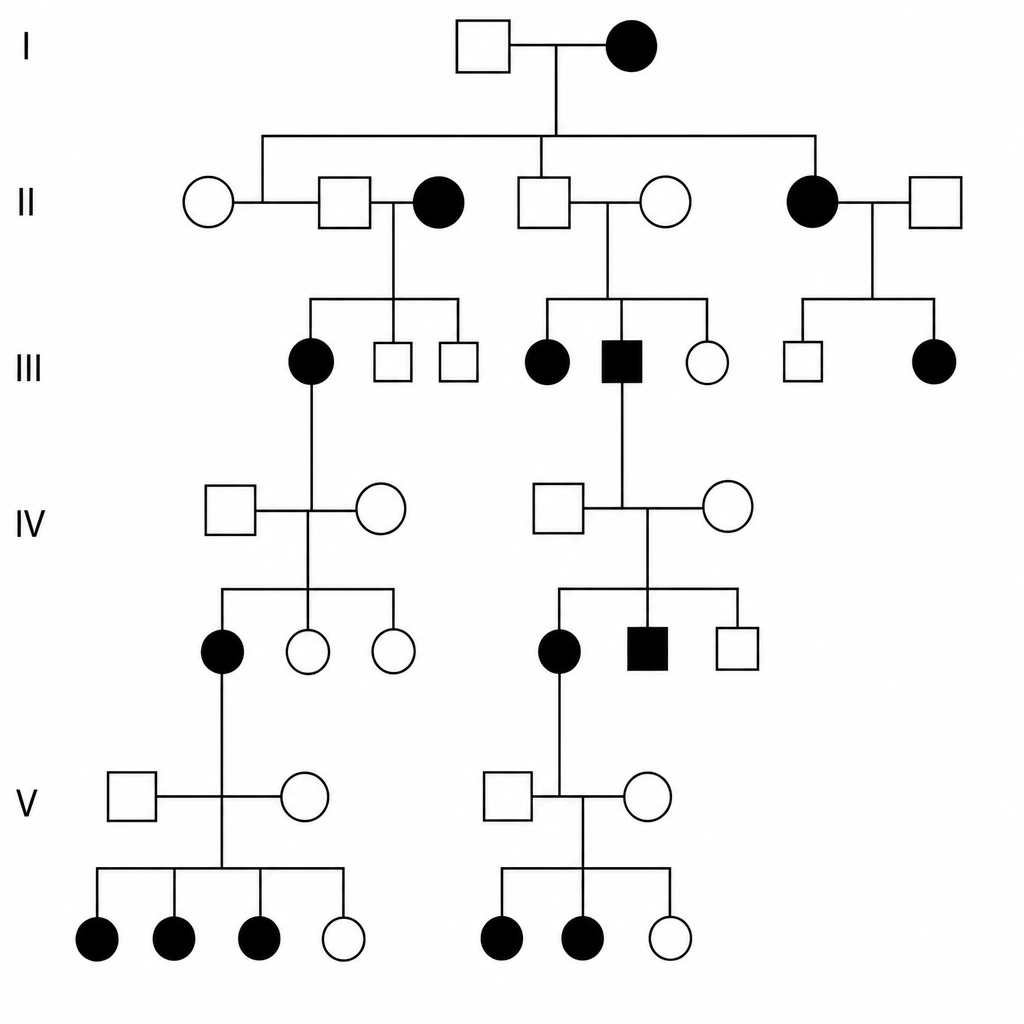
Given the pattern of inheritance shown in the pedigree, where might you find the disease gene in question?
A 31-year-old man and his wife were referred to a genetic counselor. They are concerned about the chance that their children are likely to inherit certain conditions that run in their families. The wife's father and grandfather are both healthy, but her grandfather cannot see the color red. The husband is unaware if any member of his family has the same condition. The geneticist provides some details about genetic diseases and inheritance patterns, then orders lab tests to analyze the gene mutations carried by both partners. Which of the following are the correct terms regarding the genotype and phenotype of males affected by the condition described?
A mother from rural Louisiana brings her 4-year-old son to a pediatrician. Her son is intellectually disabled, and she hopes that genetic testing will help determine the cause of her son's condition. She had previously been opposed to allowing physicians to treat her son, but his impulsive behavior and learning disabilities are making it difficult to manage his care on her own. On exam, the child has a long, thin face with a large jaw, protruding ears, and macroorchidism. The physician also hears a high-pitched holosystolic murmur at the apex of the heart that radiates to the axilla. Which of the following trinucleotide repeats is most likely affected in this individual?
A deficiency in which of the following lysosomal enzymes is inherited in a pattern similar to a deficiency of iduronate sulfatase (Hunter syndrome)?
The lac operon allows E. coli to effectively utilize lactose when it is available, and not to produce unnecessary proteins. Which of the following genes is constitutively expressed and results in the repression of the lac operon?
A healthy 29-year-old nulligravid woman comes to the physician for genetic counseling prior to conception. Her brother has a disease that has resulted in infertility, a right-sided heart, and frequent sinus and ear infections. No other family members are affected. The intended father has no history of this disease. The population prevalence of this disease is 1 in 40,000. Which of the following best represents the chance that this patient’s offspring will develop her brother's disease?
A 12-year-old boy is brought by his mother to a neurologist for continuing evaluation of seizures. His seizures were previously well-controlled on medication but over the last month he has been having seizures several times per week. The boy is non-verbal and has had severe developmental delays and cognitive disability since birth. On exam, the boy is found to be enthusiastically playing with the toys in the office and laughing at almost any stimulus. Furthermore, his movements are found to be uncoordinated with a wide based gait. Previous genetic testing has revealed an abnormality in an E3 ubiquitin ligase gene. Compared to unaffected individuals, which of the following patterns of gene expression is most likely seen in this patient?
A 16-year-old male presents to an ophthalmologist as a new patient with a complaint of blurry vision. He reports that over the past several months he has had increasing difficulty seeing the board from the back of the classroom at school. The patient is otherwise doing well in school and enjoys playing basketball. His past medical history is otherwise significant for scoliosis which is managed by an orthopedic surgeon. His family history is significant for a mother with type II diabetes mellitus, and a father who underwent aortic valve replacement last year. On physical exam, the patient is tall for his age and has long arms. He has 20 degrees of thoracic scoliosis, which is stable from previous exams. On slit-lamp examination, the patient is found to have bilateral upward lens subluxation and is prescribed corrective lenses. Which of the following is the most likely etiology of this patient’s presentation?
A 34-year-old woman comes to the fertility clinic with her husband for infertility treatment. The couple has been having unprotected intercourse for the past 2 years without any pregnancies. This is their first time seeking fertility treatment. The patient's past medical history includes asthma. She denies any menstrual irregularities, menstrual pain, abnormal bleeding or past sexually transmitted infections. The husband reports that "he would get sick easily and would always have some upper respiratory infections." Physical examination of the wife demonstrates nasal polyps bilaterally; vaginal examination is unremarkable. Physical examination of the husband is unremarkable. Semen analysis results are shown below: Semen analysis: Volume: 1.9 mL (Normal > 1.5 mL) pH: 7.4 (Normal: > 7.2) Sperm concentration: 0 million/mL (Normal: > 15 million/mL) Total sperm count: 0 million/mL (Normal: > 39 million/mL) Total motility: N/A (Normal: > 40%) Morphology: N/A (Normal: > 4% normal forms) What is the most likely explanation for this couple's infertility?
An investigator is studying the normal process of shrinking of the thymus gland with increasing age in humans. Thymic size is found to gradually start decreasing during puberty. Which of the following enzymes is most likely involved in the process underlying the decline in thymus mass with aging?
Practice by Chapter
DNA structure and organization
Practice Questions
Chromosomal structure
Practice Questions
DNA replication
Practice Questions
Cell cycle and mitosis
Practice Questions
Meiosis and genetic recombination
Practice Questions
Mutation types and consequences
Practice Questions
Mendelian inheritance patterns
Practice Questions
Non-Mendelian inheritance
Practice Questions
Genetic linkage and mapping
Practice Questions
Population genetics principles
Practice Questions
Genetic polymorphisms
Practice Questions
Gene therapy approaches
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app