
Molecular Genetics — MCQs

On this page
An adult tall male presents with a long arm span, pectus excavatum, and cardiac abnormalities. What is the most likely defective protein?
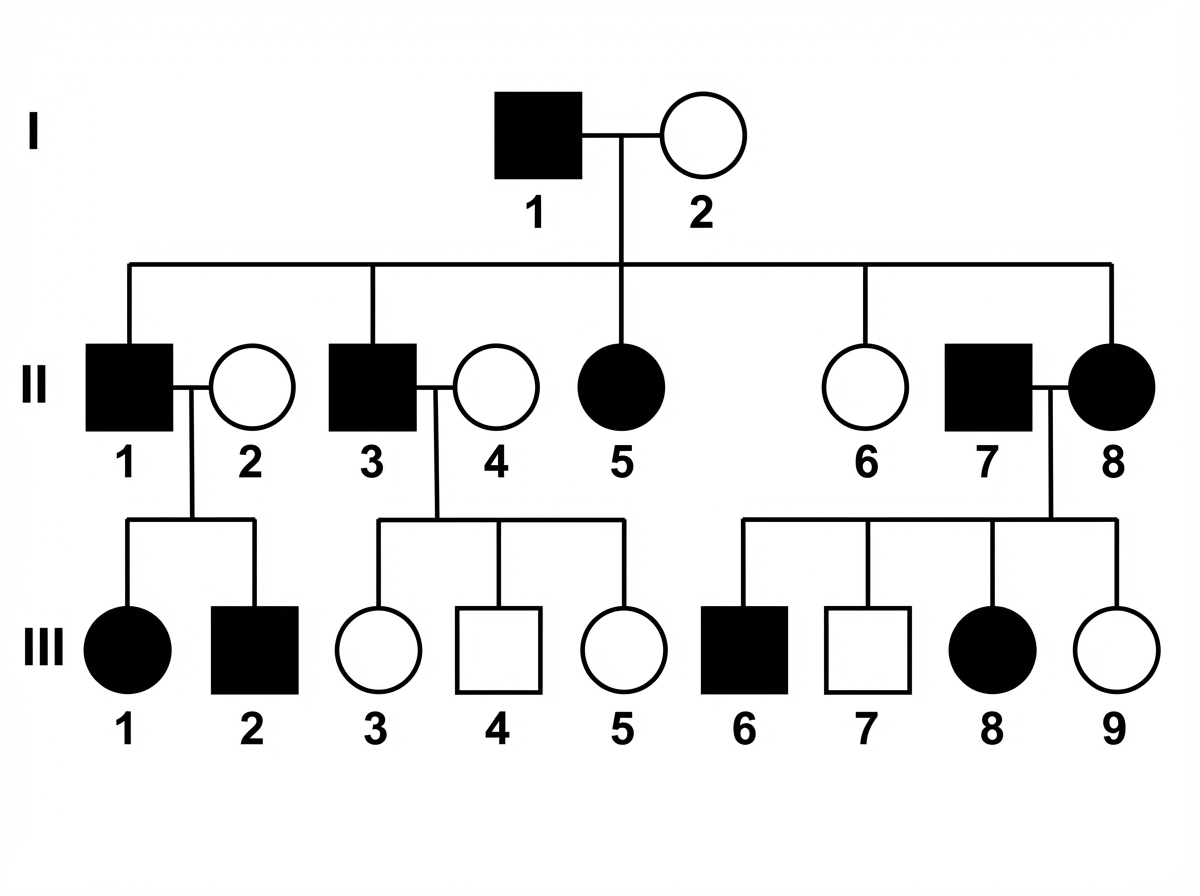
Which disease will show the mode of inheritance depicted in this pedigree?
A 12-year-old girl is brought to an oncologist, as she was recently diagnosed with a rare form of cancer. Cytogenetic studies reveal that the tumor is responsive to vinblastine, which is a cell-cycle specific anticancer agent. It acts on the M phase of the cell cycle and inhibits the growth of cells. Which of the following statements best describes the regulation of the cell cycle?
A 4-year-old male presents with a 1-year history of swaying while walking and recent episodes of tripping when ambulating. He has trouble trying to sit and get up from chairs, as well as walking up the stairs to his bedroom. On physical exam, the pediatrician notices nystagmus, absent deep tendon reflexes, significant loss of vibratory and proprioceptive sensation in his extremities, pes cavus, and slight kyphoscoliosis. A blood sample is sent for DNA sequencing and the results show a significant expansion of the trinucleotide GAA on chromosome 9. Which of the following diseases displays a similar mode of inheritance as the disease affecting this patient?
A 1-year-old child who was born outside of the United States is brought to a pediatrician for the first time because she is not gaining weight. Upon questioning, the pediatrician learns that the child has had frequent pulmonary infections since birth, and on exam the pediatrician appreciates several nasal polyps. Genetic testing is subsequently ordered to confirm the suspected diagnosis. Testing is most likely to show deletion of which of the following amino acids from the protein involved in this child's condition?
A 19-year-old man presents to the office for a routine physical exam and a meningitis vaccination prior to attending college on a basketball scholarship. Also present at the appointment is his father who appears to be in his mid-sixties and is much shorter. The patient’s pulse is 70/min, respirations are 18/min, temperature is 37.0°C (98.6°F), and blood pressure is 120/80 mm Hg. He is 183 cm (6 ft 0 in) tall and weighs 79.4 kg (175 lb). His heart rate is regular with a mild diastolic murmur (II/VI) over the aortic valve and his lungs are clear to auscultation bilaterally. A scoliosis test shows mild deviation of his thoracic spine. A skin examination shows numerous red-to-white linear markings on the skin around his lower back. His fingers are long. Which of the following genes does this patient most likely have a mutation of?
An 11-year-old boy is brought to the physician for the evaluation of frequent falling. His mother reports that the patient has had increased difficulty walking over the last few months and has refused to eat solid foods for the past 2 weeks. He has met all developmental milestones. The patient has had multiple ear infections since birth. His temperature is 37°C (98.6°F), pulse is 90/min, and blood pressure is 120/80 mm Hg. Examination shows foot inversion with hammertoes bilaterally. His gait is wide-based with irregular and uneven steps. Laboratory studies show a serum glucose concentration of 300 mg/dL. Further evaluation of this patient is most likely to show which of the following findings?
A 9-year-old boy is admitted to the hospital for placement of halo gravitational traction in order to treat his previously observed kyphoscoliosis. Specifically, he has a previously diagnosed curve that has gotten worse over time and now threatens to compromise his thoracic cavity. His past medical history is significant for short stature, and he has consistently been below the 5th percentile for height since birth. On physical exam, he is found to have macrocephaly with frontal bossing, short arms and legs with disproportionate shortening of the proximal segments, and lumbar lordosis. Which of the following proteins are most likely mutated in this patient?
A mother brings her son to the pediatrician because she is concerned about his health. She states that throughout her child's life he has demonstrated aggressive behavior. However, he has recently begun biting himself causing injury and bleeding. The patient has a past medical history of mental retardation and episodes of severe joint pain. His temperature is 99.5°F (37.5°C), blood pressure is 87/48 mmHg, pulse is 90/min, respirations are 17/min, and oxygen saturation is 98% on room air. Physical exam reveals a child attempting to bite his arms. Which of the following is the inheritance pattern of the disease with which this patient presents?
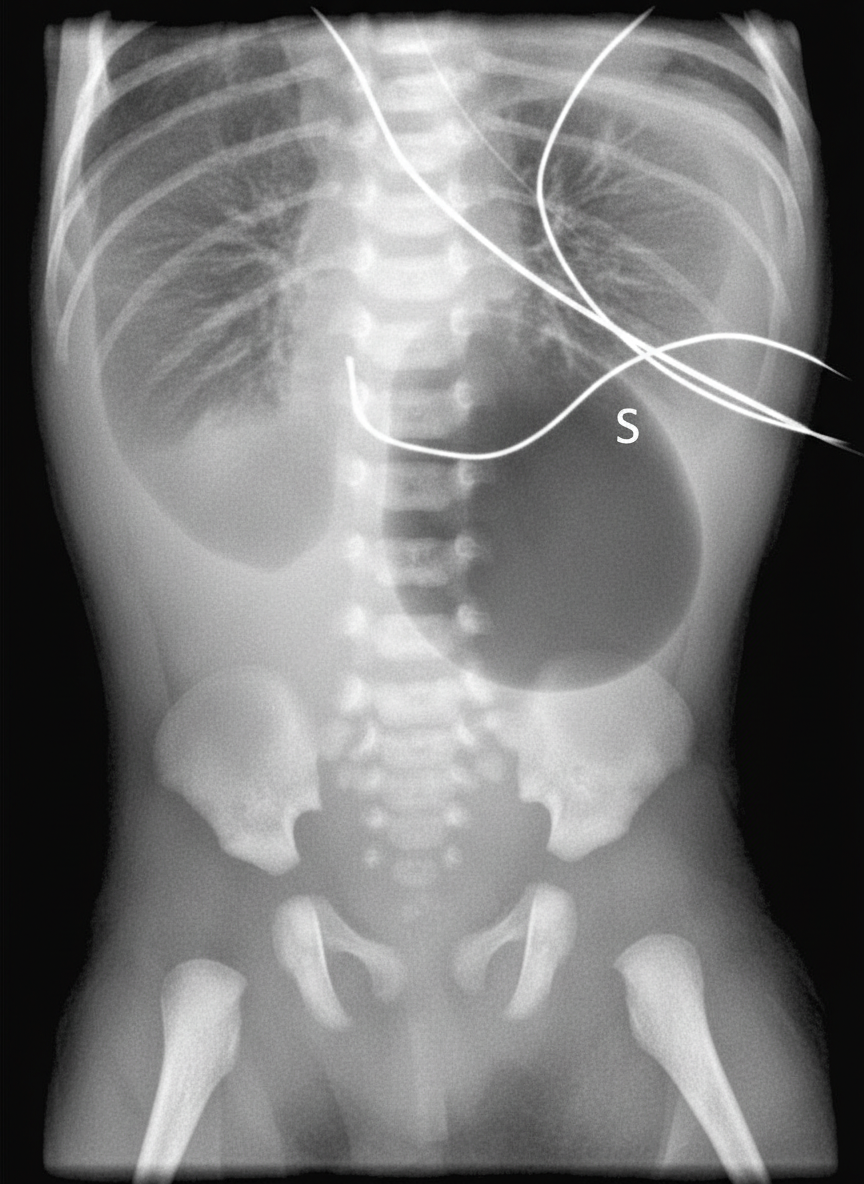
A 40-year-old woman brings her 2-day-old infant to the pediatrician’s office for a routine checkup. She tells the pediatrician that her baby vomits a greenish-yellow fluid after every feeding session. She has not been very successful in feeding him due to this problem. She also says that her baby has not passed stool since they left the hospital. On examination, the pediatrician observes that the baby has a flat facial profile and small eyes. The epicanthal folds are prominent and the palms have a single transverse crease. His abdomen is distended with high-pitched bowel sounds. The pediatrician orders an abdominal radiograph, the film is shown in the picture. Which of the following best explains the physical and clinical features exhibited by this infant?
Practice by Chapter
DNA structure and organization
Practice Questions
Chromosomal structure
Practice Questions
DNA replication
Practice Questions
Cell cycle and mitosis
Practice Questions
Meiosis and genetic recombination
Practice Questions
Mutation types and consequences
Practice Questions
Mendelian inheritance patterns
Practice Questions
Non-Mendelian inheritance
Practice Questions
Genetic linkage and mapping
Practice Questions
Population genetics principles
Practice Questions
Genetic polymorphisms
Practice Questions
Gene therapy approaches
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app