
Metabolism — MCQs

On this page
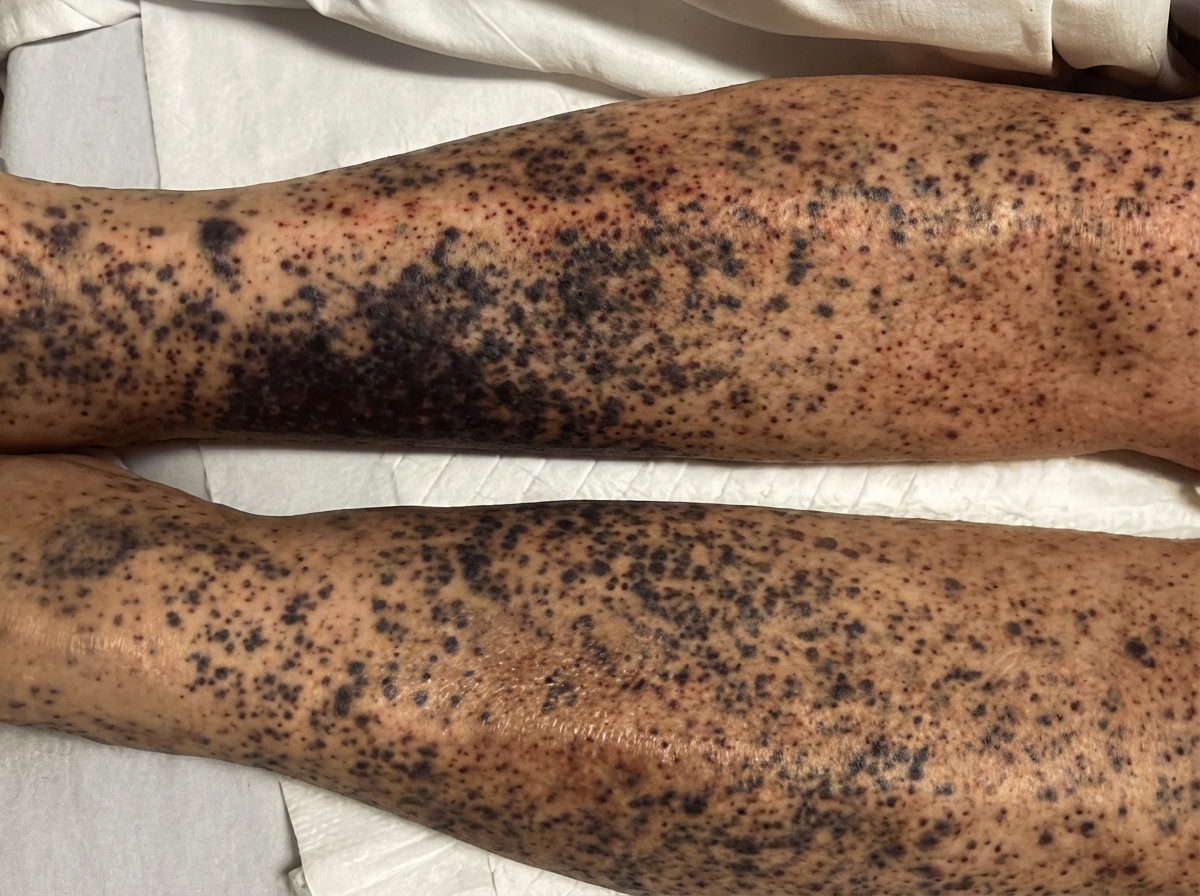
A 59-year-old man is brought to the emergency department with signs of spontaneous bruising of the lower legs. The patient has a history of alcohol use disorder and has been unemployed for the last 2 years. He reports a 1-year history of fatigue and joint pain. Physical examination of the patient’s legs reveals the findings illustrated in the image. Oral examination shows swollen gums, petechiae of the hard palate, and poor dentition. The most likely underlying cause of this patient's current findings involves which of the following metabolic deficiencies?
A 24-year-old man presents to the emergency department complaining of a prolonged course of diarrhea. He reports that he has had 3–4 large volume watery stools daily for the last several weeks. He has no pain with bowel movements, no abdominal pain, and no blood in his stools. He is homeless and uses recreational drugs. He also reports that he usually drinks a half-liter of whiskey, or whatever else he can find, every day and he has done this for several years. The physical exam is notable for a hyperpigmented rash across his face, neck, chest, and the backs of his hands and forearms. On mental status exam, he is oriented to person and place but not time; he scores a 23/30 on the Montreal Cognitive Assessment (MOCA). This patient's presentation is most likely related to which of the following micronutrients?
A 6-month-old boy is brought to the emergency department by his mother because of recurrent vomiting and yellowing of his eyes. The mother says that he has been eating poorly since she started weaning him off of breast milk 5 days ago. At this time, mashed vegetables and fruits were added to his diet. Examination shows scleral jaundice and dry mucous membranes. The tip of the liver is palpable 4 cm below the right costal margin. His serum glucose concentration is 47 mg/dL, serum alanine aminotransferase is 55 U/L, and serum aspartate aminotransferase is 66 U/L. Which of the following enzymes is most likely deficient?
A 32-year-old woman presents with dysuria, urinary frequency, and suprapubic discomfort. She developed these symptoms about a week ago, which was 5 days after she had finished treatment with ceftriaxone for otitis media. She has a single sexual partner and uses oral contraceptives. She is allergic to macrolides, azoles, and nystatin. Her vital signs are as follows: blood pressure is 110/60 mm Hg, heart rate is 80/min, respiratory rate is 15/min, and temperature is 36.6℃ (97.9℉). Examination reveals findings consistent with an uncomplicated urinary tract infection. Considering the spectrum of agents she is allergic to, she is prescribed a sulfonamide, a competitive inhibitor against an important bacterial enzyme. Which of the following Michaelis-Menten plots describes the kinetics of condensation of para-aminobenzoic acid with dihydropterin pyrophosphate by dihydropteroate synthase under the influence of sulfanilamide?
A 2-year-old boy is brought to the emergency department by his mother 30 minutes after having a generalized tonic-clonic seizure. He was born at home and has not attended any well-child visits. He is not yet able to walk and does not use recognizable words. His paternal uncle has a severe intellectual disability and has been living in an assisted-living facility all of his life. The boy's urine phenylacetate level is markedly elevated. Which of the following amino acids is most likely nutritionally essential for this patient because of his underlying condition?
A 4-month-old boy is brought to the physician because of a seizure. He was delivered at term after an uncomplicated pregnancy. He is currently at the 10th percentile for height, 5th percentile for weight, and 15th percentile for head circumference. Examination shows muscle hypotonia. His serum lactic acid and alanine are elevated. A functional assay of pyruvate dehydrogenase complex in serum leukocytes shows decreased enzyme activity. Supplementation with which of the following substances should be avoided in this patient?
A 3-month-old boy is brought to the pediatrician by his mother after she notices orange sand–like crystalline material in her child’s diaper. He is not currently taking any medication and is exclusively breastfed. His immunizations are up to date. The doctor tells the mother that her son may have an X-linked recessive disorder. The boy is prescribed a medication that inhibits an enzyme responsible for the production of the crystals seen in his urine microscopy. Which of the following enzymes is the target of this medication?
You have isolated cells from a patient with an unknown disorder and would like to locate the defect in this patient. When radiolabeled propionate is added to the mitochondria, no radiolabeled carbon dioxide is detected. However, when radiolabeled methylmalonic acid is added, radiolabeled carbon dioxide is detected from these cells. Which of the following amino acids can be fully metabolized by this patient?
A 53-year-old homeless woman is brought to the emergency department by the police after she was found in the park lying unconscious on the ground. Both of her pupils are normal in size and reactive to light. There are no signs of head trauma. Finger prick test shows a blood glucose level of 20 mg/dL. She has been brought to the emergency department for acute alcohol intoxication several times before. Her vitals signs include: blood pressure 100/70 mm Hg, heart rate 90/min, respiratory rate 22/min, and temperature 35.0℃ (95.0℉). On general examination, she looks pale, but there is no sign of icterus noted. On physical examination, the abdomen is soft and non-tender and no hepatosplenomegaly noted. She spontaneously opens her eyes after the administration of a bolus of intravenous dextrose, thiamine, and naloxone. Blood and urine samples are drawn for toxicology screening. Finally, the blood alcohol level turns out to be 300 mg/dL. What will be the most likely laboratory findings in this patient?
A 25-year-old woman presents her physician with a complaint of feeling tired and low on energy for the past 6 months. She also has noticed she has been having trouble performing daily tasks and at times experiencing near-fainting spells. She has no recollection of similar instances in the past. Her past medical history is insignificant, except for the fact that she has been a strict vegan for the last 5 years. Her vital signs are stable. On physical examination, she is visibly pale and has decreased position and vibratory sensation in her both lower extremities. There is decreased lower limb reflexes with sensation intact. A complete blood count - done last week, - shows hemoglobin of 9.7 g/dL with an MCV of 110 fL. The serum levels of which of the following will most likely aid in the physician’s treatment plan?
Practice by Chapter
TCA cycle reactions and regulation
Practice Questions
Electron transport chain and oxidative phosphorylation
Practice Questions
Pentose phosphate pathway
Practice Questions
Gluconeogenesis
Practice Questions
Glycogen metabolism
Practice Questions
Amino acid metabolism
Practice Questions
Integration of metabolic pathways
Practice Questions
Fed state vs. fasting state metabolism
Practice Questions
Exercise metabolism
Practice Questions
Alcohol metabolism
Practice Questions
Metabolic adaptations in starvation
Practice Questions
Metabolic disorders overview
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app