
Metabolism — MCQs

On this page
A 38-year-old man presents to a fertility specialist. He is concerned that he is infertile. His wife had two children from a previous marriage and has regular menses. They have been married three years and have been trying to conceive for the past two. His vitals are normal. Physical exam reveals bilateral gynecomastia, elongated limbs, and small testicles. Levels of plasma gonadotropins are elevated. Which of the following is likely to be also elevated in this patient?
A 44-year-old woman is brought to the emergency department because of confusion and agitation. She was brought by police after she was found walking along a highway. The patient's brother comes to join her soon after her arrival. He says she has peptic ulcer disease and hypertension. He thinks she drinks around half a bottle of vodka daily. Her current medications include omeprazole and hydrochlorothiazide, although the brother is unsure if she takes them regularly. Her temperature is 37.1°C (98.7°F), pulse is 90/min, respirations are 16/min, and blood pressure is 135/90 mm Hg. On mental status examination, she is confused and not oriented to person, place, or time. Neurologic examination shows horizontal nystagmus. Her gait is wide-based with small steps. Her hemoglobin concentration is 9 g/dL. Her serum homocysteine concentration is elevated and her methylmalonic acid concentration is within the reference range. A peripheral blood smear shows hypersegmented neutrophils. Which of the following is the most likely cause of this patient's anemia?
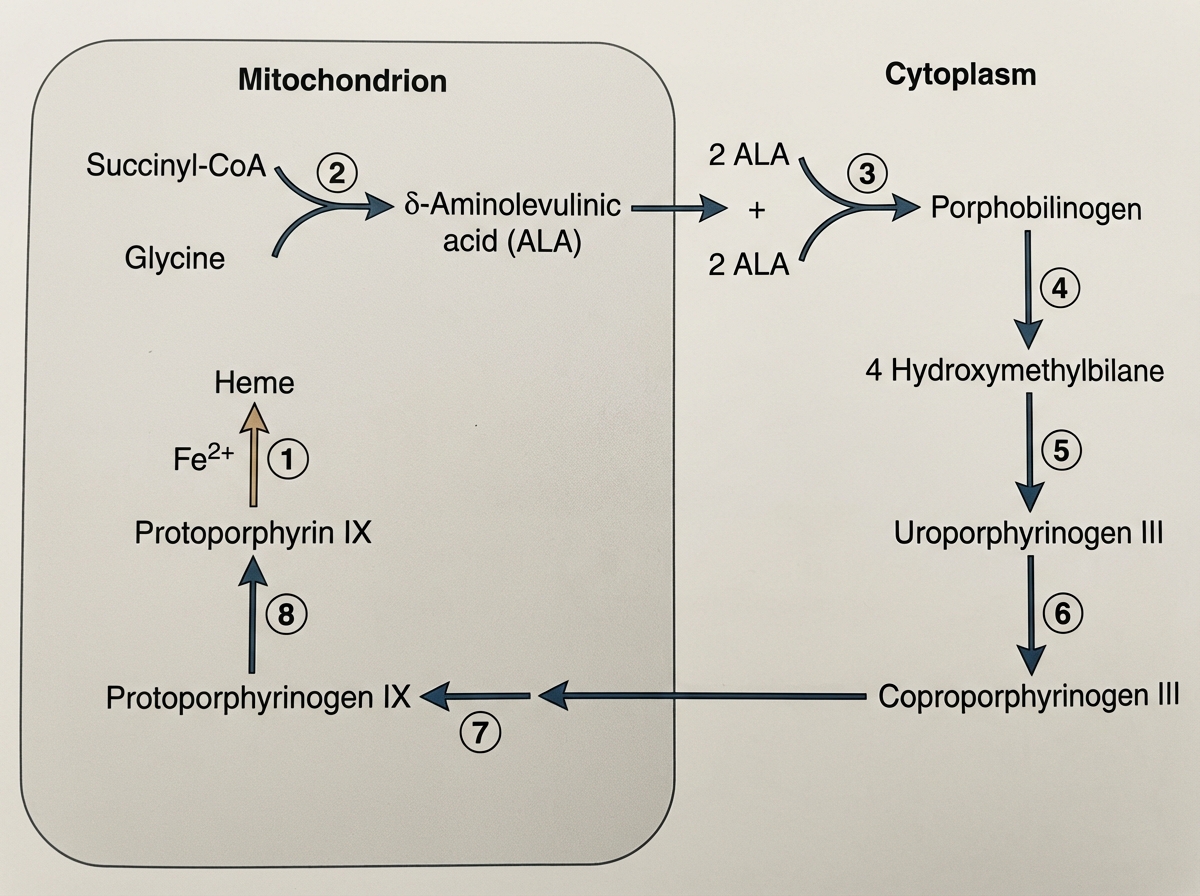
A family who recently moved from Nebraska to Texas visits the pediatrician. They have a 3-year-old child that had been developing normally before this change in location. The child became lethargic, fatigued, pale, and constipated 3 months after moving to the new house. Also, the blood smear of the patient demonstrates the finding of sideroblasts. Analyze the scheme presented below. Which of the following enzymes labeled as no. 1 is impaired in this patient and causing his symptoms?
A 6-day-old female newborn is brought to the physician because of yellowish discoloration of her eyes and body, vomiting, and poor feeding for 3 days. She has had diarrhea for the past 2 days. She was born at 38 weeks' gestation and the antenatal period was uncomplicated. She appears lethargic. Vital signs are within normal limits. Examination shows jaundice of the skin and conjunctivae. Bilateral cataracts are present. The abdomen is soft and nontender. The liver is palpated 4-cm below the right costal margin; there is no splenomegaly. Muscle tone is decreased in all extremities. Serum glucose concentration is 37 mg/dL. Which of the following is the most appropriate recommendation to prevent long-term complications of this illness?
A 10-month-old boy is brought to his pediatrician because of a 3-day history of fever and lethargy. He has previously had more infections than expected since birth but otherwise appears to be developing normally. On exam, the boy is found to have a purulent, erythematous bump on his left upper extremity. This lesion is cultured and found to have a catalase-positive, coagulase-positive, gram-positive organism, which is the same organism that caused his previous infections. Based on clinical suspicion, a nitroblue tetrazolium (NBT) test is obtained that confirms the diagnosis. The substrate of the protein that is most likely defective in this patient is produced by which of the following metabolic pathways?
A 3-day-old infant presents because the patient’s parents noticed that his skin was becoming yellow. The mother said that the patient eats well, has normal stool and urine color. It’s her first child from first healthy pregnancy. The patient was born on time and delivered via spontaneous vaginal delivery with no complications. Family history is significant for a maternal aunt who died as an infant of unknown causes. The patient is afebrile and vital signs are within normal limits. On physical examination, he is awake, calm, and looks healthy, except for the yellow tone of the skin and scleral icterus. Laboratory findings are significant for elevated unconjugated bilirubin, with a normal complete blood count. Other routine laboratory blood tests are within normal limits. The patient is treated with phototherapy, but his jaundice worsens and his unconjugated hyperbilirubinemia persists well into the second week of life. Which of the following is the most likely diagnosis in this patient?
While studying vesicular trafficking in mammalian epithelial cells, a scientist identified a specific protein that was responsible for contorting the plasma membrane to capture extracellular materials and forming endosomes. This protein also helps transport those endosomes from the trans-Golgi network to lysosomes. Which of the following is the protein that the scientists identified?
A 2-month-old boy is brought to the pediatrician by his parents after they notice that he had a "floppy" appearance, poor suckling, vomiting, and spontaneous generalized movements a few weeks after birth. The boy was born at home, and routine newborn screening was normal. On physical examination, the infant is hypotonic, has poor suckling, cannot hold his head straight while prone, and does not follow objects. He has fair skin, red hair, blue eyes, and eczema. At the second appointment, laboratory tests show high levels of phenylalanine and prolactin and low levels of homovanillic acid and serotonin. Which of the following enzymes is deficient in this patient?
A 43-year-old woman presents to a hematology clinic to discuss the results of a bone marrow biopsy that was performed about 4 weeks ago. She was referred to this clinic to evaluate her chronic anemia after all other noninvasive diagnostic testing was inconclusive. Today her blood pressure is 114/76 mm Hg, pulse is 94/min, respiratory rate 21/min, and temperature is 36.6°C (97.9°F). She has mild jaundice and shortness of breath. The bone marrow aspirate showed erythroid precursors with multiple cytoplasmic structures that were highlighted with a Prussian blue stain. A deficiency of which of the following would result in these findings?
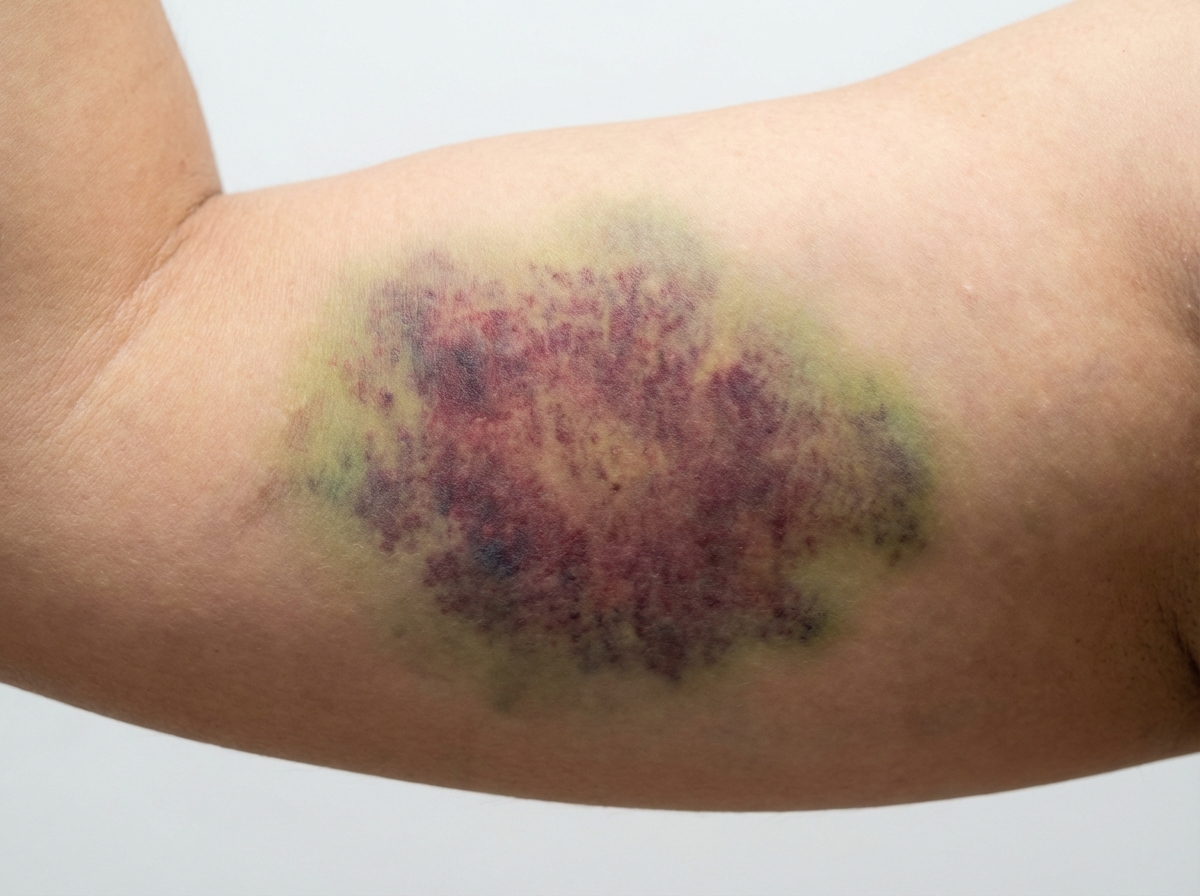
A 27-year-old man comes to the physician for a follow-up evaluation. Two days ago, he was involved in a physical altercation and sustained a bruise on his left arm and an injury to his left shoulder. Initially, there was a reddish-purple discoloration on his left upper arm. A photograph of the left upper arm today is shown. Which of the following enzymes is most likely responsible for the observed changes in color?
Practice by Chapter
TCA cycle reactions and regulation
Practice Questions
Electron transport chain and oxidative phosphorylation
Practice Questions
Pentose phosphate pathway
Practice Questions
Gluconeogenesis
Practice Questions
Glycogen metabolism
Practice Questions
Amino acid metabolism
Practice Questions
Integration of metabolic pathways
Practice Questions
Fed state vs. fasting state metabolism
Practice Questions
Exercise metabolism
Practice Questions
Alcohol metabolism
Practice Questions
Metabolic adaptations in starvation
Practice Questions
Metabolic disorders overview
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app