
Metabolism — MCQs

On this page
A 27-year-old man presents to the emergency department with painless yellowing of his skin. The patient states he is generally healthy and has no past medical history. He smokes 2 packs of cigarettes per day and was recently treated for a urinary tract infection with a single dose of ceftriaxone followed by a 7 day course of ciprofloxacin. He recently returned from a 3 day hiking trip and is an avid vegan. His only other medical history is a mild cough for the past few days. His temperature is 97.5°F (36.4°C), blood pressure is 122/82 mmHg, pulse is 85/min, respirations are 15/min, and oxygen saturation is 98% on room air. Physical exam reveals an abdomen which is non-tender. Mild scleral icterus and sublingual jaundice is noted. Which of the following is the most likely etiology of this patient’s symptoms?
An 8-year-old boy is referred to your office by his school for kyphoscoliosis. His mother recently noticed a change in the way he walks but thought it was a normal part of his growth. She notes that he has always been clumsy and has frequent falls. He has a history of type 1 diabetes mellitus for which he receives insulin. He has no other health problems and has been doing well in school. On physical exam his temperature is 99°F (37.2°C), blood pressure is 110/75 mmHg, pulse is 80/min, and respirations are 19/min. Cardiopulmonary exam is unremarkable. On neurologic exam you notice nystagmus. Patellar reflex is absent and the patient has a staggering gait. The disorder most likely responsible for this patient’s presentation is due to an abnormality in which of the following?
A 3-month-old infant is brought to her pediatrician for a well-child visit. The infant was born to a 22-year-old mother via a spontaneous vaginal delivery at 38 weeks of gestation in her home. She moved to the United States approximately 3 weeks ago from a small village. She reports that her infant had 2 episodes of non-bloody and non-bilious vomiting. The infant's medical history includes eczema and 2 seizure episodes that resolved with benzodiazepines in the emergency department. Physical examination is notable for a musty body odor, eczema, and a fair skin complexion. Which of the following is the best next step in management?
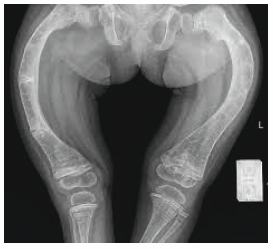
A 6-year-old boy is brought to the physician for a well-child examination. His mother has noticed he frequently falls while running. He was born at term and pregnancy was uncomplicated. He has a seizure disorder treated with phenytoin. He is at the 20th percentile for height and at 30th percentile for weight. Vital signs are within normal limits. Examination shows decreased muscle strength in the lower extremities. There is a deep groove below the costal margins bilaterally. An x-ray of the lower extremities is shown. Which of the following is the most likely cause of these findings?
A 59-year-old man is brought to the physician by his wife for a psychiatric evaluation. Over the past 12 months, his behavior has become increasingly disruptive. His wife no longer brings him along shopping because he has attempted to grope a female cashier on 2 occasions. He has begun to address the mail carrier using a racial epithet. Three years later, the patient dies. Light microscopy of sections of the frontal and temporal lobes shows intracellular inclusions of transactive response DNA binding protein (TDP-43). These proteins are bound to a regulatory molecule that usually marks them for degradation. The regulatory molecule in question is most likely which of the following?
A 45-year-old man is brought to the emergency department by ambulance after vomiting blood. The patient reports that he only ate a small snack the morning before and had not eaten anything for over 24 hours. At the hospital, the patient is stabilized. He is admitted to a surgical floor and placed on NPO with a nasogastric tube set to intermittent suction. He has been previously diagnosed with liver cirrhosis. An esophagogastroduodenoscopy (EGD) has been planned for the next afternoon. At the time of endoscopy, some pathways were generating glucose to maintain serum glucose levels. Which of the following enzymes catalyzes the irreversible biochemical reaction of this process?
In cells with impaired production of reducing factors for respiratory burst, decreased production of which substance is most likely observed?
A 31-year-old male comes to the physician because of a 2-day history of blisters and brownish discoloration of urine. His symptoms appeared after he returned from a 4-day trip with his friends in Florida. He has had similar episodes of blistering twice in the past three years. Each episode resolved spontaneously after a few weeks. Examination shows vesicles and bullae on the face and the dorsal surfaces of his hands and forearms. His condition is most likely caused by a defect in which of the following enzymes?
A 9-year-old boy is brought to his primary care physician after his mom noticed that he was limping. He says that he has been experiencing significant hip and knee pain over the last 2 months but thought he may have just strained a muscle. Radiographs show a collapse of the femoral head, and he is diagnosed with Legg-Calve-Perthes disease. He undergoes surgery and is placed in a Petrie cast from his hips to his toes bilaterally so that he is unable to move his knees or ankles. Eight weeks later, the cast is removed, and he is found to have significantly smaller calves than before the cast was placed. Which process in myocytes is most likely responsible for this finding?
A 12-year-old girl is brought to the physician because of a 2-hour history of severe epigastric pain, nausea, and vomiting. Her father has a history of similar episodes of abdominal pain and developed diabetes mellitus at the age of 30 years. Abdominal examination shows guarding and rigidity. Ultrasonography of the abdomen shows diffuse enlargement of the pancreas; no gallstones are visualized. Which of the following is the most likely underlying cause of this patient's condition?
Practice by Chapter
TCA cycle reactions and regulation
Practice Questions
Electron transport chain and oxidative phosphorylation
Practice Questions
Pentose phosphate pathway
Practice Questions
Gluconeogenesis
Practice Questions
Glycogen metabolism
Practice Questions
Amino acid metabolism
Practice Questions
Integration of metabolic pathways
Practice Questions
Fed state vs. fasting state metabolism
Practice Questions
Exercise metabolism
Practice Questions
Alcohol metabolism
Practice Questions
Metabolic adaptations in starvation
Practice Questions
Metabolic disorders overview
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app