
Metabolism — MCQs

On this page
A 5-year-old boy is brought to the physician’s office with complaints of being tired constantly, which has limited his ability to walk or play with his friends. Physical examination in the physician’s office is normal. Further testing reveals that the patient has a genetic mutation in an enzyme and muscle biopsy shows high levels of alpha-ketoglutarate and low levels of succinyl-CoA as compared to normal. The enzyme that is most likely deficient in this patient requires which of the following as a cofactor?
A 26-year-old African American man comes to the physician because of a 3-day history of fatigue, back pain, and dark urine. One week ago, he developed a headache and was treated with aspirin. He does not smoke or use illicit drugs. Physical examination shows conjunctival pallor. A peripheral blood smear shows erythrocytes with inclusions of denatured hemoglobin. Which of the following enzymes is involved in providing precursors for nucleotide synthesis in this patient?
An 8-day-old boy is brought to the physician by his mother because of vomiting and poor feeding. The pregnancy was uncomplicated, and he was born at full term. He appears pale and lethargic. Physical examination shows diffusely increased muscle tone. His urine is noted to have a sweet odor. This patient's symptoms are most likely caused by the accumulation of which of the following?
A 6-year-old boy is brought to the office by his mother. She reports that her son is well but has some concerns about his overall health: he is shorter and, physically, seems less developed compared to his siblings when they were the same age. He recently started school and the mother reports that the boy’s teachers are concerned with his learning capability. His height and weight are in the 10th and 15th percentiles, respectively. Lab results reveal: Hemoglobin 10 gm/dL Mean corpuscular volume 110 fL Multi-segmented neutrophils are seen on peripheral blood smear. Urinary orotic acid levels are found to be high. What is the most likely cause of this patient’s condition?
Parkinson’s disease is a progressive neurodegenerative disease. It is characterized by a loss of dopaminergic neurons in the substantia nigra pars compacta and the formation of cellular inclusions called Lewy bodies. These are composed of α-synuclein that has been bound to ubiquitin. In healthy individuals, α-synuclein bound to ubiquitin would be degraded by which of the following?
A 5-month-old boy presents with increasing weakness for the past 3 months. The patient’s mother says that the weakness is accompanied by dizziness, sweating, and vertigo early in the morning. Physical examination shows hepatomegaly. Laboratory findings show an increased amount of lactate, uric acid, and elevated triglyceride levels. Which of the following enzymes is most likely deficient in this patient?
A 12-year-old girl comes to the clinic with a grossly enlarged abdomen. She has a history of frequent episodes of weakness, sweating, and pallor that are eliminated by eating. Her development has been slow. She started to walk unassisted at 2 years and was not performing well at school. Physical examination reveals a blood pressure of 100/60 mm Hg, heart rate of 80/min, and temperature of 36.9°C (98.4℉). On physical examination, the liver is enlarged, firm, and palpable up to the pelvis. The spleen and kidney are not palpable. Laboratory investigation reveals low blood glucose and pH with high lactate, triglycerides, ketones, and free fatty acids. The liver biopsy revealed high glycogen content. Hepatic glycogen structure was normal. The enzyme assay performed on the biopsy tissue revealed very low glucose-6-phosphatase levels. What is the most likely diagnosis?
A 3-year-old African-American female presents to the emergency department with fatigue. Her parents endorse malaise and weakness on behalf of the patient for two weeks. Her temperature is 98.9°F (37.2°C), blood pressure is 94/70 mmHg, pulse is 102/min, and respirations are 22/min. On physical exam, she is tired-appearing with conjunctival pallor. Her parents report that they immigrated from Liberia before the patient was born. They deny any family history of medical disorders, and the patient has no sick contacts at home. Laboratory tests are performed and reveal the following: Leukocyte count: 10,700/mm^3 Hemoglobin: 8.6 g/dL Hematocrit: 24% Mean corpuscular volume: 84 µm^3 Platelet count: 488,000/mm^3 Reticulocyte index: 3.8% The patient should receive which of the following nutritional supplements?
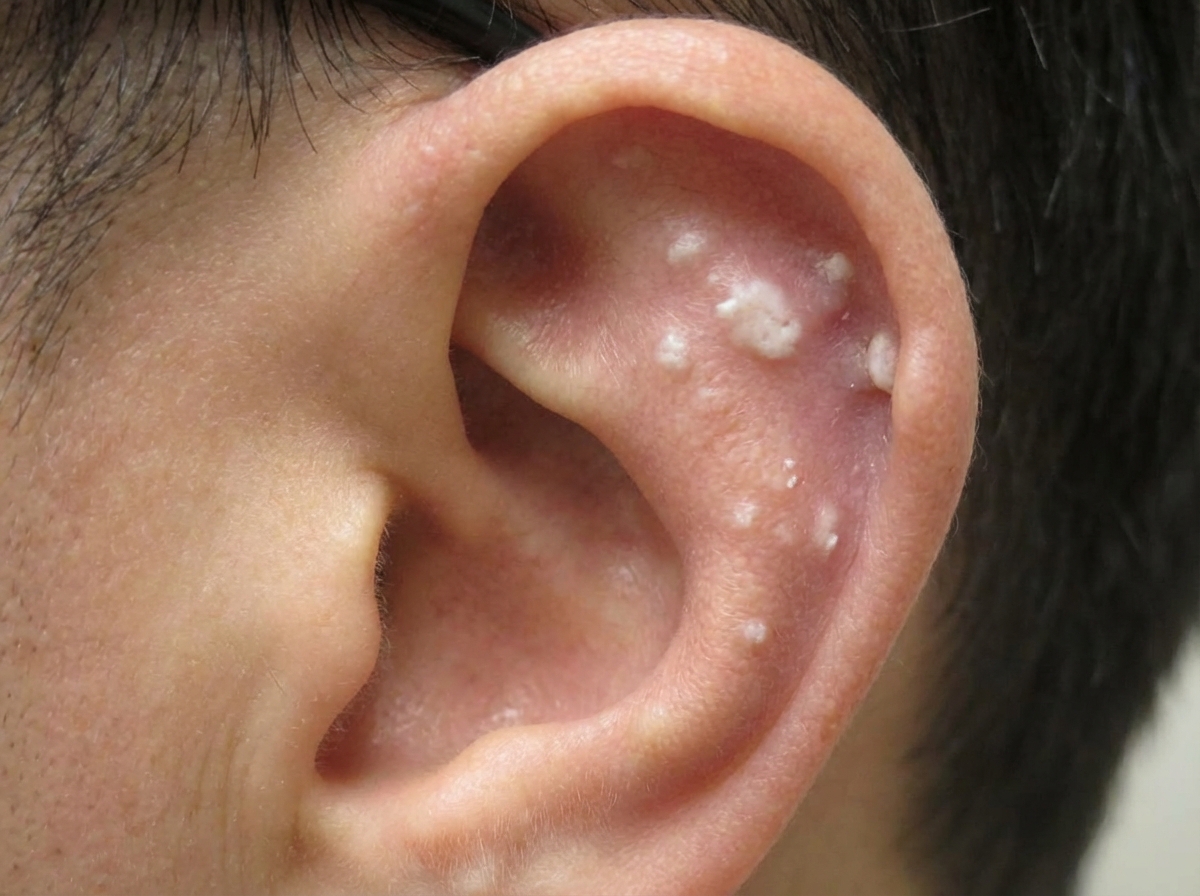
A 43-year-old man comes to the physician because of left flank pain and nausea for 2 hours. The pain comes in waves and radiates to his groin. Over the past year, he has had intermittent pain in the bilateral flanks and recurrent joint pain in the toes, ankles, and fingers. He has not seen a physician in over 10 years. He takes no medications. He drinks 3–5 beers daily. His sister has rheumatoid arthritis. Vital signs are within normal limits. Physical examination shows marked tenderness bilaterally in the costovertebral areas. A photograph of the patient's left ear is shown. A CT scan of the abdomen shows multiple small kidney stones and a 7-mm left distal ureteral stone. A biopsy of the patient's external ear findings is most likely to show which of the following?
A 17-year-old girl presents to her primary care physician for a wellness checkup. The patient is currently doing well in school and plays soccer. She has a past medical history of childhood obesity that was treated with diet and exercise. The patient states that her menses have not changed, and they occur every 1 to 3 months. Her temperature is 99.5°F (37.5°C), blood pressure is 127/70 mmHg, pulse is 90/min, respirations are 13/min, and oxygen saturation is 98% on room air. The patient's BMI at this visit is 22.1 kg/m^2. On physical exam, the patient is in no distress. You note acne present on her face, shoulders, and chest. You also note thick, black hair on her upper lip and chest. The patient's laboratory values are seen as below. Hemoglobin: 14 g/dL Hematocrit: 42% Leukocyte count: 7,500/mm^3 with normal differential Platelet count: 177,000/mm^3 Serum: Na+: 137 mEq/L Cl-: 101 mEq/L K+: 4.4 mEq/L HCO3-: 24 mEq/L BUN: 27 mg/dL Glucose: 90 mg/dL Creatinine: 1.0 mg/dL Ca2+: 10.1 mg/dL Testosterone: 82 ng/dL 17-hydroxyprogesterone: elevated AST: 12 U/L ALT: 10 U/L Which of the following is associated with this patient's most likely diagnosis?
Practice by Chapter
TCA cycle reactions and regulation
Practice Questions
Electron transport chain and oxidative phosphorylation
Practice Questions
Pentose phosphate pathway
Practice Questions
Gluconeogenesis
Practice Questions
Glycogen metabolism
Practice Questions
Amino acid metabolism
Practice Questions
Integration of metabolic pathways
Practice Questions
Fed state vs. fasting state metabolism
Practice Questions
Exercise metabolism
Practice Questions
Alcohol metabolism
Practice Questions
Metabolic adaptations in starvation
Practice Questions
Metabolic disorders overview
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app