
Metabolism — MCQs

On this page
Which of the following foods should be consumed to prevent thiamine deficiency?
A patient with homocystinuria presents with ectopia lentis (dislocation of the lens). Which vitamin should be supplemented?
What is the primary function of IL-8?
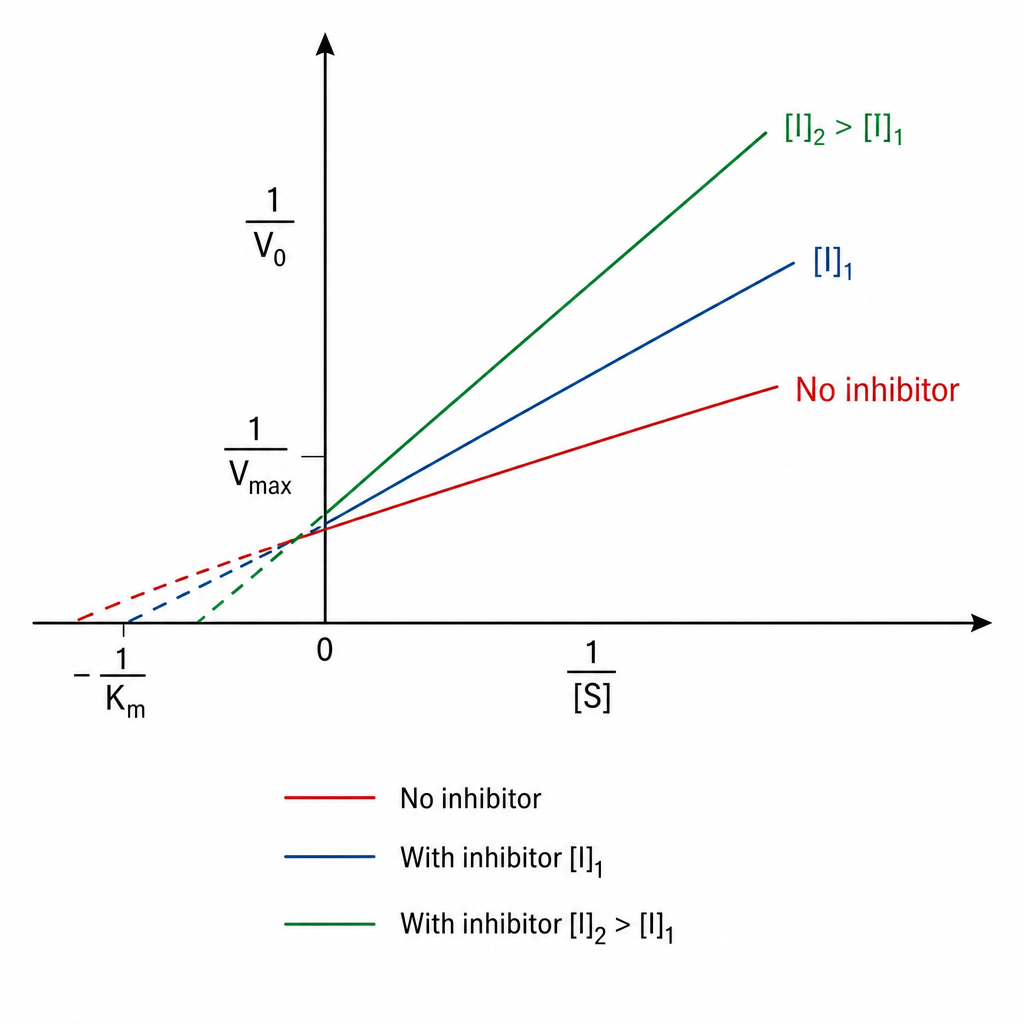
Identify the type of inhibition exhibited by A?
A 45-year-old patient presents with joint pain and weakness and is known to have homocystinuria. Which vitamin is required in the treatment?
Practice by Chapter
TCA cycle reactions and regulation
Practice Questions
Electron transport chain and oxidative phosphorylation
Practice Questions
Pentose phosphate pathway
Practice Questions
Gluconeogenesis
Practice Questions
Glycogen metabolism
Practice Questions
Amino acid metabolism
Practice Questions
Integration of metabolic pathways
Practice Questions
Fed state vs. fasting state metabolism
Practice Questions
Exercise metabolism
Practice Questions
Alcohol metabolism
Practice Questions
Metabolic adaptations in starvation
Practice Questions
Metabolic disorders overview
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app