
Sphingolipidoses (Tay-Sachs, Gaucher, Niemann-Pick) — MCQs

An 8-month-old boy is brought to the physician by his parents for gradually increasing loss of neck control and inability to roll over for the past 2 months. During this time, he has had multiple episodes of unresponsiveness with a blank stare and fluttering of the eyelids. His parents state that he sometimes does not turn when called but gets startled by loud noises. He does not maintain eye contact. He was able to roll over from front to back at 5 months of age and has not yet begun to sit or crawl. His parents are of Ashkenazi Jewish descent. Neurological examination shows generalized hypotonia. Deep tendon reflexes are 3+ bilaterally. Plantar reflex shows extensor response bilaterally. Fundoscopy shows bright red macular spots bilaterally. The remainder of the examination shows no abnormalities. Which of the following is the most likely cause of this patient's symptoms?
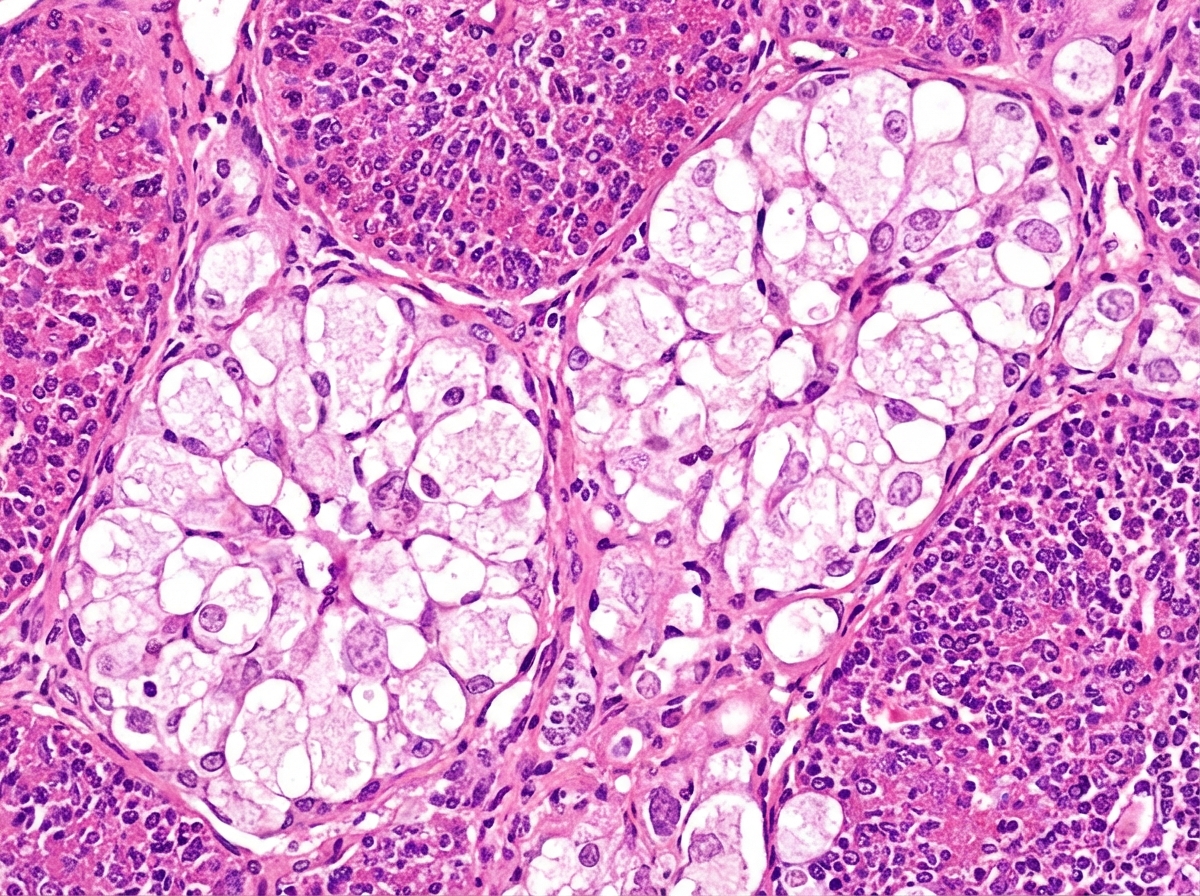
An 18-month-old boy is brought to the physician because of a 2-day history of cough, fever, and lethargy. He has been admitted to the hospital twice during the past year for pneumonia. He can stand without support but has not started to walk. He speaks in bisyllables. He is at the 3rd percentile for height and 4th percentile for weight. Examination shows diffuse crackles over bilateral lung fields. Abdominal examination shows hepatosplenomegaly. Fundoscopy shows bright red macular spots. Despite being given appropriate antibiotic therapy, the patient dies. A photomicrograph of a section of the spleen obtained during autopsy is shown. Accumulation of which of the following substances is the most likely cause of this patient's condition?
A 2-year-old boy is brought to the emergency department by his parents because of fever and recurrent episodes of jerky movements of his extremities for the past 6 hours. Pregnancy and delivery were uncomplicated, and development was normal until the age of 1 year. The parents report that he has had gradual loss of speech, vision, and motor skills over the past year. During this time, he has been admitted to the hospital three times because of myoclonic seizures. Physical examination shows hypertonicity of the upper and lower extremities. Fundoscopic examination shows pallor of the optic disc bilaterally. An MRI of the brain shows brain atrophy and hyperintensity of the periventricular and subcortical areas. Two days after admission, the patient dies. Histopathologic examination of the brain shows aggregation of globoid cells and loss of glial cells. The patient’s condition was most likely caused by a deficiency of which of the following enzymes?
An 18-month-old boy of Ashkenazi-Jewish descent presents with loss of developmental milestones. On ocular exam, a cherry-red macular spot is observed. No hepatomegaly is observed on physical exam. Microscopic exam shows lysosomes with onion-skin appearance. What is the most likely underlying biochemical abnormality?
An 8-month-old female infant from a first-degree consanguineous couple was brought to the physician because the mother noticed abnormalities in the growth of her child as well as the different lengths of her child's legs. The infant had gingival hyperplasia, restricted movement in both shoulders, a prominent, pointed forehead, and enophthalmos with a slight opacity in both corneas. A blood test revealed 10 fold higher than normal levels of the following enzymes: N-acetyl-ß-glucosaminidase, ß-glucuronidase, ß-hexosaminidase A, and alkaline phosphatase. Which of the following is most likely deficient in this patient?
A 4-month-old male infant is brought in because he rejects food and is losing weight. He had several upper respiratory tract infections during the last 2 months. Upon examination, hepatosplenomegaly is noted, as well as mild hypotonia. During the next few weeks, hepatosplenomegaly progresses, the boy fails to thrive, and he continues to reject food. He has a blood pressure of 100/70 mm Hg and heart rate of 84/min. Blood tests show pancytopenia and elevated levels of transaminases. Slit lamp examination shows bilateral cherry-red spots on the macula. Chest X-ray shows a reticulonodular pattern and calcified nodules. Biopsy of the liver shows foamy histiocytes. What is the most likely diagnosis?
A 6-month-old infant male is brought to the emergency department with a 1-hour history of vomiting and convulsions. He was born at home and had sporadic prenatal care though his parents say that he appeared healthy at birth. He initially fed well; however, his parents have noticed that he has been feeding poorly and is very irritable since they moved on to baby foods. They have also noticed mild yellowing of his skin but assumed it would go away over time. On presentation, he is found to be very sleepy, and physical exam reveals an enlarged liver and spleen. The rest of the physical exam is normal. Which of the following enzymes is most likely functioning abnormally in this patient?
A 5-year-old girl is brought in for a routine checkup. She was born at 39 weeks gestation via spontaneous vaginal delivery and is up to date on all vaccines and is meeting all developmental milestones. Upon examination, she is pale with a few petechiae on her chest neck and back. Examination of the abdomen reveals painless hepatosplenomegaly. Liver enzymes are mildly elevated and complete blood cell count shows slight anemia and thrombocytopenia. Iron, B12, and folate are normal. A bone marrow biopsy shows mildly hypocellular marrows with diffuse macrophages with eosinophilic cytoplasm. The cytoplasm looks like wrinkled tissue paper on further inspection. No blasts are observed. What is the most likely diagnosis in the present case?
A 26-year-old woman presents to a physician for genetic counseling, because she is worried about trying to have a child. Specifically, she had 2 siblings that died young from a lysosomal storage disorder and is afraid that her own children will have the same disorder. Her background is Ashkenazi Jewish, but she says that her husband's background is mixed European heritage. Her physician says that since her partner is not of Jewish background, their chance of having a child with Niemann-Pick disease is dramatically decreased. Which of the following genetic principles best explains why there is an increased prevalence of this disease in some populations?
A research study evaluates three siblings with Niemann-Pick disease type C: a 6-year-old with ataxia and vertical supranuclear gaze palsy, a 10-year-old with hepatosplenomegaly and mild cognitive impairment, and a 14-year-old who is asymptomatic. Genetic testing reveals all three carry the same compound heterozygous NPC1 mutations. Fibroblast studies show similar cholesterol esterification defects and filipin staining patterns. Miglustat therapy is available. Evaluate the biological basis for phenotypic variability and optimal treatment allocation.
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app