
Diagnostic approaches — MCQs

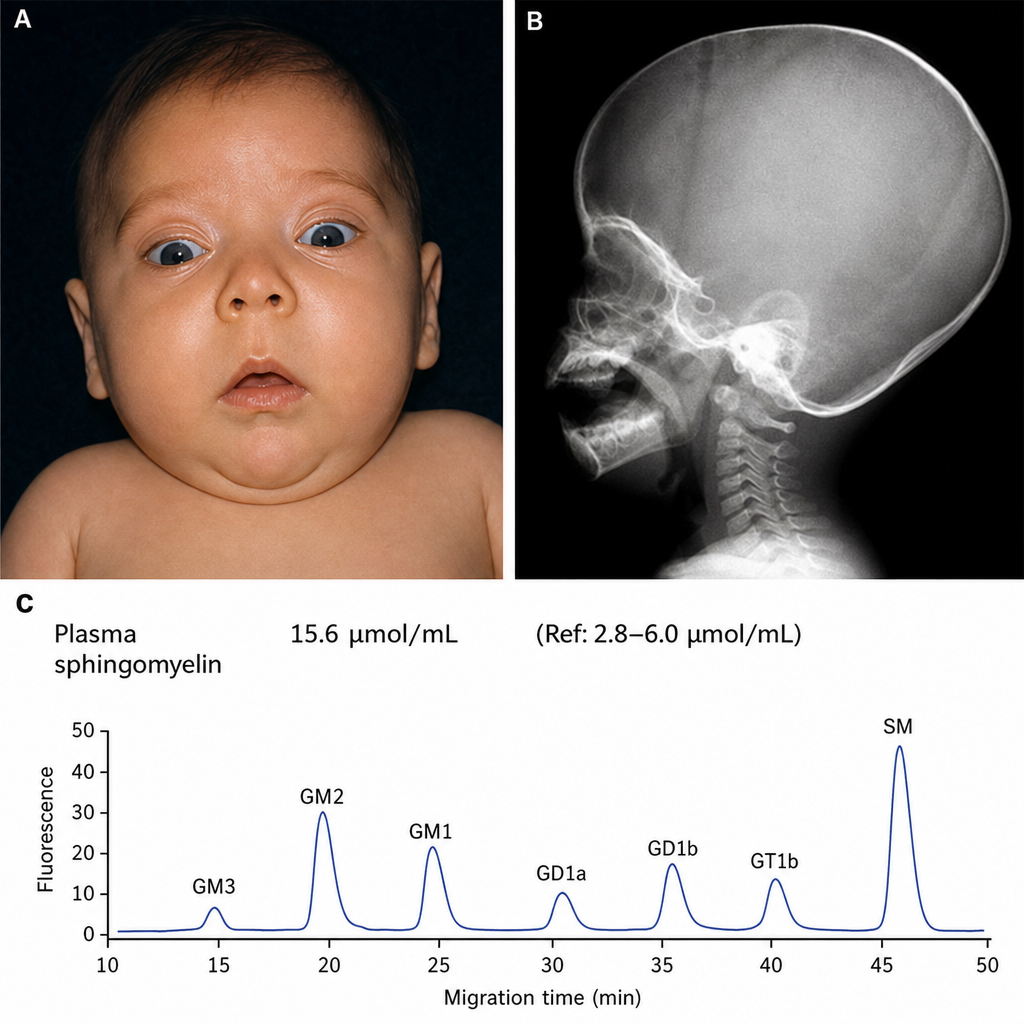
You examine an infant in your office. On exam you observe hypotonia, as well as the findings shown in Figures A and B. You order laboratory testing, which demonstrates the findings shown in Figure C. Which of the following is the most likely pathologic mechanism involved?
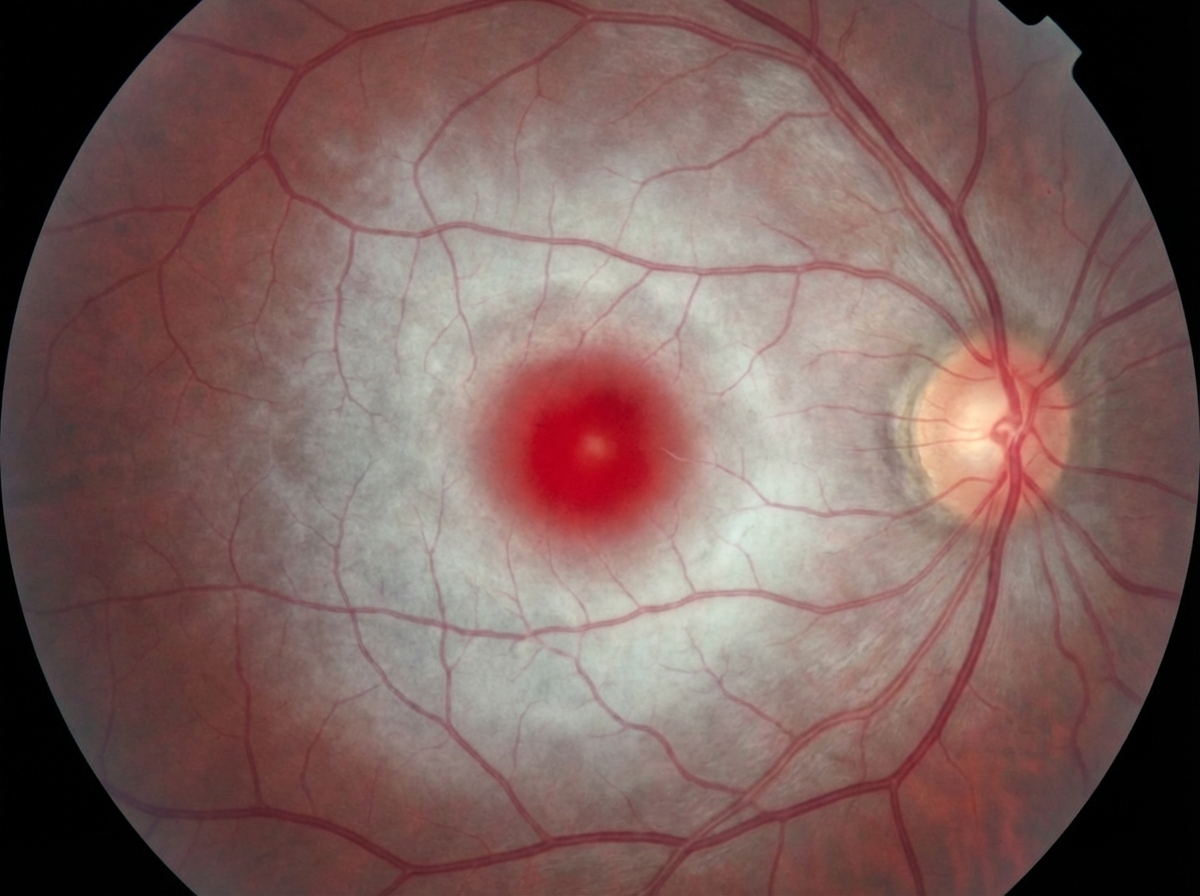
A 7-month-old boy is brought by his parents to the pediatrician’s office. His mother says the child has been weakening progressively and is not as active as he used to be when he was born. His condition seems to be getting worse, especially over the last month. He was born at 41 weeks through normal vaginal delivery. There were no complications observed during the prenatal period. He was progressing well over the 1st few months and achieving the appropriate milestones. On examination, his abdomen appears soft with no liver enlargement. The patient appears to be dehydrated and lethargic. The results of a fundoscopic examination are shown in the picture. A blood test for which of the following enzymes is the next best assay to evaluate this patient's health?
A deficiency in which of the following lysosomal enzymes is inherited in a pattern similar to a deficiency of iduronate sulfatase (Hunter syndrome)?
A 37-year-old primigravid woman comes to the physician at 13 weeks' gestation for a prenatal visit. She feels well. Her only medication is folic acid. Vital signs are within normal limits. Pelvic examination shows a uterus consistent in size with a 13-week gestation. Ultrasonography shows a nuchal translucency above the 99th percentile. Maternal serum pregnancy-associated plasma protein A is decreased and human chorionic gonadotropin concentrations are elevated to 2 times the median level. Which of the following is most likely to confirm the diagnosis?
A 4-month-old male infant is brought in because he rejects food and is losing weight. He had several upper respiratory tract infections during the last 2 months. Upon examination, hepatosplenomegaly is noted, as well as mild hypotonia. During the next few weeks, hepatosplenomegaly progresses, the boy fails to thrive, and he continues to reject food. He has a blood pressure of 100/70 mm Hg and heart rate of 84/min. Blood tests show pancytopenia and elevated levels of transaminases. Slit lamp examination shows bilateral cherry-red spots on the macula. Chest X-ray shows a reticulonodular pattern and calcified nodules. Biopsy of the liver shows foamy histiocytes. What is the most likely diagnosis?
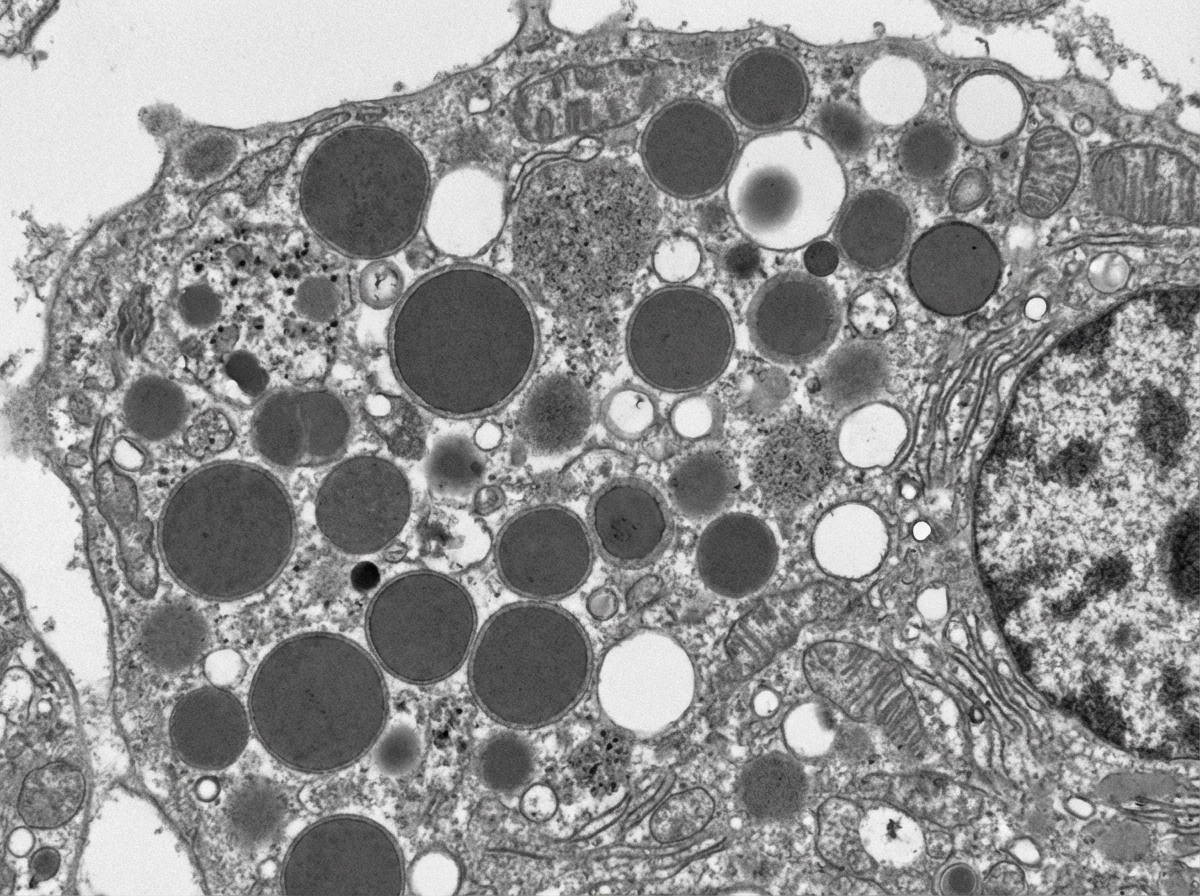
A 3-month-old boy presents for routine health maintenance. The patient has coarse facial features and stiff joint movements with restricted passive and active range of motion. He also has problems following objects with his eyes and seems not to focus on anything. On physical examination, the corneas are clouded, and the patient fails to meet any 3-month developmental milestones. Genetic testing and histopathology are performed and reveal failure of a cellular structure to phosphorylate mannose residues on glycoproteins. An electron microscopy image of one of this patient’s cells is shown. Which of the following is the most likely diagnosis in this patient?
A 26-year-old woman presents to a physician for genetic counseling, because she is worried about trying to have a child. Specifically, she had 2 siblings that died young from a lysosomal storage disorder and is afraid that her own children will have the same disorder. Her background is Ashkenazi Jewish, but she says that her husband's background is mixed European heritage. Her physician says that since her partner is not of Jewish background, their chance of having a child with Niemann-Pick disease is dramatically decreased. Which of the following genetic principles best explains why there is an increased prevalence of this disease in some populations?
A 3-month-old infant is brought to her pediatrician for a well-child visit. The infant was born to a 22-year-old mother via a spontaneous vaginal delivery at 38 weeks of gestation in her home. She moved to the United States approximately 3 weeks ago from a small village. She reports that her infant had 2 episodes of non-bloody and non-bilious vomiting. The infant's medical history includes eczema and 2 seizure episodes that resolved with benzodiazepines in the emergency department. Physical examination is notable for a musty body odor, eczema, and a fair skin complexion. Which of the following is the best next step in management?
A research study evaluates three siblings with Niemann-Pick disease type C: a 6-year-old with ataxia and vertical supranuclear gaze palsy, a 10-year-old with hepatosplenomegaly and mild cognitive impairment, and a 14-year-old who is asymptomatic. Genetic testing reveals all three carry the same compound heterozygous NPC1 mutations. Fibroblast studies show similar cholesterol esterification defects and filipin staining patterns. Miglustat therapy is available. Evaluate the biological basis for phenotypic variability and optimal treatment allocation.
A 15-year-old boy with Hunter syndrome (MPS II) on weekly enzyme replacement therapy develops IgG antibodies with high neutralizing capacity against idursulfase. His symptoms have worsened over the past 6 months with increasing hepatosplenomegaly and joint stiffness. His brother with the same mutation shows excellent response to ERT without antibody formation. Synthesize an appropriate management plan considering immunologic and genetic factors.
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app