
Lipid metabolism — MCQs

On this page
A 6-month-old infant male is brought to the emergency department with a 1-hour history of vomiting and convulsions. He was born at home and had sporadic prenatal care though his parents say that he appeared healthy at birth. He initially fed well; however, his parents have noticed that he has been feeding poorly and is very irritable since they moved on to baby foods. They have also noticed mild yellowing of his skin but assumed it would go away over time. On presentation, he is found to be very sleepy, and physical exam reveals an enlarged liver and spleen. The rest of the physical exam is normal. Which of the following enzymes is most likely functioning abnormally in this patient?
A 14-year-old male of eastern European descent presents to the free clinic at a university hospital for a respiratory infection, which his mother explains occurs quite frequently. The male is noted to be of short stature, have a gargoyle-like facies, clouded corneas, poor dentition, and is severely mentally retarded. A urinalysis revealed large amounts of heparan and dermatan sulfate. Which of the following is the most likely diagnosis?
An autopsy was performed on a 2-year-old male child. The clinical report stated that the child's parents were first cousins, and that he had deteriorated physically and mentally over the past year, becoming deaf, unable to eat, and paralyzed. A brain biopsy demonstrated the accumulation of GM2-gangliosides in the neurons. Which of the following enzymes was missing from this child?
A 3-month-old girl is brought to the emergency department in respiratory distress after her parents noticed that she was having difficulty breathing. They say that she developed a fever 2 days ago and subsequently developed increasing respiratory difficulty, lethargy, and productive cough. On presentation, her temperature is 103°F (39.5°C), blood pressure is 84/58 mmHg, pulse is 141/min, and respirations are 48/min. Physical exam reveals subcostal retractions and consolidation in the right lower lung field. She is also found to have coarse facial features and restricted joint movement. Serum laboratory tests reveal abnormally elevated levels of lysosomal enzymes circulating in the blood. The enzyme that is most likely defective in this patient has which of the following substrates?
A 2-year-old girl presents to the emergency room with a witnessed seizure-like episode characterized by bilateral arm twitching, foaming at the mouth, and unresponsiveness lasting 30 seconds. The mother reports recent lethargy and current antibiotic treatment for an ear infection, but denies fever, pain, trauma, feeding changes, or gastrointestinal symptoms. Examination is unremarkable. Lab results show a glucose level of 1.9 mmol/L (34 mg/dL). What is the most likely cause of the patient's seizure?
A 3-year-old boy is brought in to his pediatrician by his mother after she noticed that the child was starting to turn yellow. She has not noticed any behavioral changes. On exam, the boy is icteric but is behaving normally. His temperature is 98.8°F (37.1°C), blood pressure is 108/78 mmHg, pulse is 78/min, and respirations are 14/min. His labs are drawn, and he is found to have an unconjugated hyperbilirubinemia with a serum bilirubin of 15 mg/dL. The mother is counseled that this boy's condition may require phenobarbital as a treatment to reduce his bilirubin levels. Which of the following is the most likely defect in this child?
A 14-year-old Caucasian male patient found to have low serum copper, high urine copper, and low serum ceruloplasmin is placed on penicillamine for management of his genetic disorder. Which of the following is LEAST consistent with this patient's clinical picture?
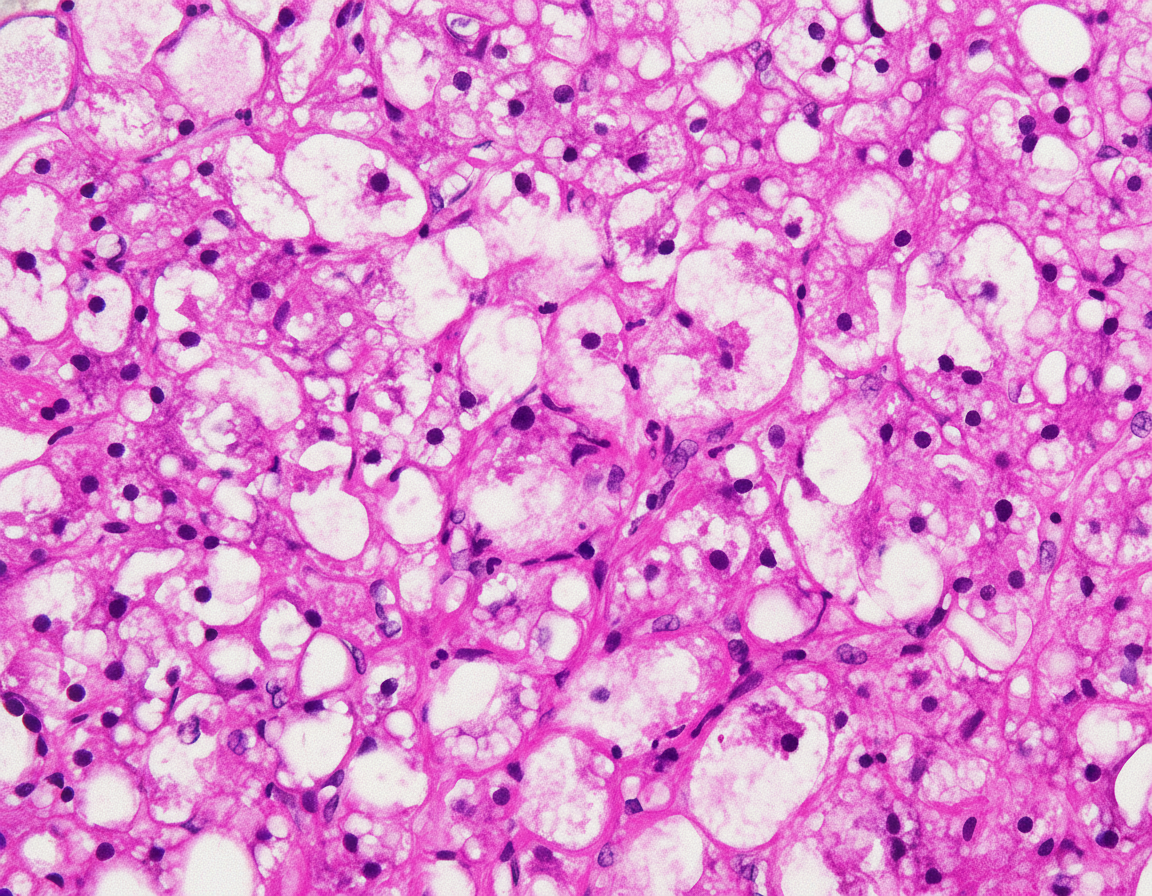
A 43-year-old woman comes to the physician because of a 2-week history of malaise, nausea, and a 3-kg (6.6-lb) weight loss. She has been drinking 8–9 alcoholic beverages daily for the past 20 years. Her temperature is 37.8°C (100°F) and pulse is 105/min. Examination shows jaundice and hepatosplenomegaly. A photomicrograph of a section of a biopsy specimen of the liver is shown. Which of the following mechanisms best explains the findings shown?
A 17-year-old female is found to have an inherited deficiency of alpha-galactosidase A. Skin biopsy shows accumulation of ceramide trihexose in the tissue. Which of the following abnormalities would be expected in this patient?
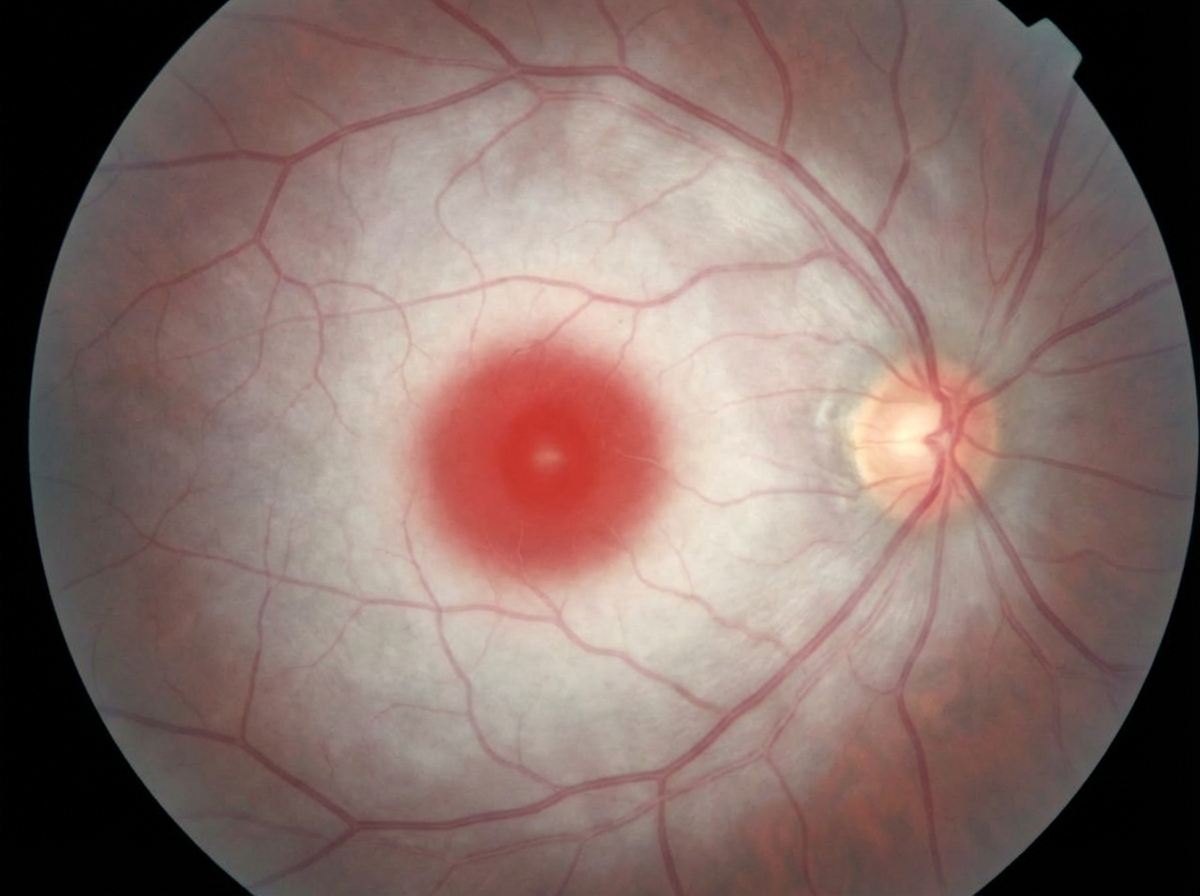
A 7-month-old boy is brought to the pediatrician by his parents due to progressively worsening weakness for the last three months. The parents also describe the boy as having an exaggerated response when startled as well as diminishing response to visual stimuli. At birth, the boy was healthy and remained as such for the first few months of life. The mother says pregnancy was unremarkable, and the boy was born at 39 weeks with no complications during delivery. He is up to date on his vaccinations. The boy's grandparents immigrated from an eastern European country. Physical examination reveals hyperreflexia. Abdominal examination reveals no abnormalities. On fundoscopy, the following is seen. Which of the following is most likely deficient in this patient?
Practice by Chapter
Fatty acid oxidation (beta-oxidation)
Practice Questions
Fatty acid synthesis
Practice Questions
Ketone body metabolism
Practice Questions
Cholesterol synthesis and regulation
Practice Questions
Lipoprotein metabolism
Practice Questions
Phospholipid metabolism
Practice Questions
Eicosanoid synthesis and function
Practice Questions
Steroid hormone synthesis
Practice Questions
Adipose tissue metabolism
Practice Questions
Brown vs. white adipose tissue
Practice Questions
Disorders of lipid metabolism
Practice Questions
Integration with carbohydrate metabolism
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app