
Lipid metabolism — MCQs

On this page
A 17-year-old girl is brought to the physician because she has never menstruated. She is at the 15th percentile for weight and 45th percentile for height. Vital signs are within normal limits. Examination shows facial hair, clitoromegaly, and coarse, curly pubic hair that extends to the inner surface of both thighs. She has no glandular breast tissue. Ultrasound shows inguinal testes but no uterus or ovaries. Which of the following is the most likely underlying cause for this patient's symptoms?
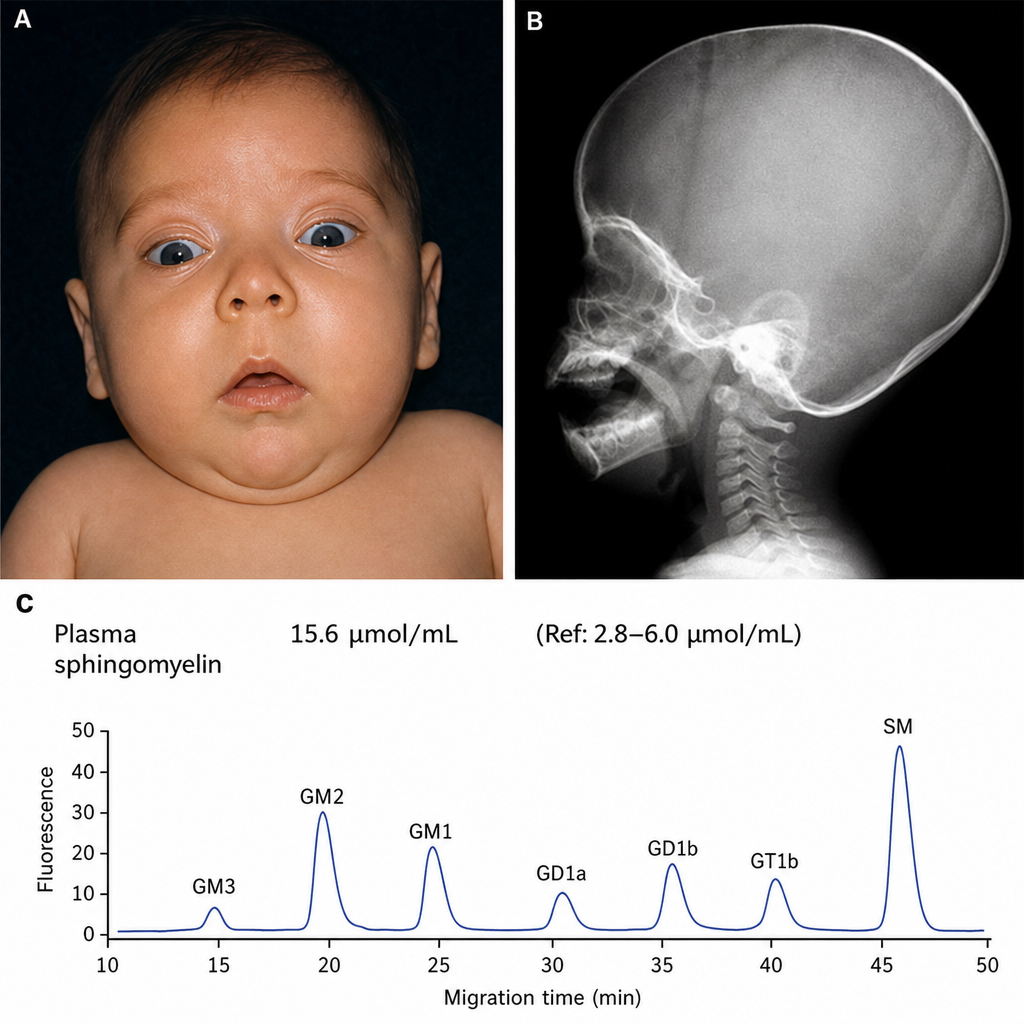
You examine an infant in your office. On exam you observe hypotonia, as well as the findings shown in Figures A and B. You order laboratory testing, which demonstrates the findings shown in Figure C. Which of the following is the most likely pathologic mechanism involved?
A 10-month-old boy with a seizure disorder is brought to the physician by his mother because of a 2-day history of vomiting and lethargy. Laboratory studies show a decreased serum glucose concentration with low ketones. Further testing confirms a deficiency in an enzyme involved in fatty acid oxidation. Which of the following enzymes is most likely deficient in this patient?
A 38-year-old woman presents to her surgeon 1 year after a surgery for Crohn disease involving the removal of much of her small bowel. She had no major complications during the surgery and recovered as expected. Since then, she has noticed bone pain and weakness throughout her body. She has also had several fractures since the surgery. A panel of labs relevant to bone physiology was obtained and the results are shown below: Serum: Phosphate: Decreased Calcium: Decreased Alkaline phosphatase: Increased The factor that is most likely abnormal in this patient can also be synthesized from which of the following?
A 9-month-old girl is brought to the physician by her parents for multiple episodes of unresponsiveness in which she stares blankly and her eyelids flutter. She has gradually lost control of her neck and ability to roll over during the past 2 months. She is startled by loud noises and does not maintain eye contact. Her parents are of Ashkenazi Jewish descent. Neurological examination shows generalized hypotonia. Deep tendon reflexes are 3+ bilaterally. Fundoscopy shows bright red macular spots bilaterally. Abdominal examination shows no abnormalities. Which of the following metabolites is most likely to accumulate due to this patient's disease?
A 1-year-old male is found to have high blood pressure on multiple visits to your office. On examination, the patient has normal genitalia. Further laboratory workup reveals low serum aldosterone and high serum testosterone. Which of the following is most likely to be elevated in this patient?
A newborn is delivered at term to a 38-year-old woman after an uncomplicated pregnancy and delivery. The newborn's blood pressure is 142/85 mm Hg. Examination shows clitoral enlargement and labioscrotal fusion. Serum studies show a sodium of 151 mg/dL and a potassium of 3.2 mg/dL. Karyotype analysis shows a 46, XX karyotype. The patient is most likely deficient in an enzyme that is normally responsible for which of the following reactions?
A 28-year-old man presents to the emergency department with diffuse abdominal pain and nausea for the past 5 hours. The pain started with a dull ache but is now quite severe. He notes that he “just doesn’t feel like eating” and has not eaten anything for almost a day. Although the nausea is getting worse, the patient has not vomited. He notes no medical issues in the past and is not currently taking any medications. He admits to drinking alcohol (at least 2–3 bottles of beer per day after work and frequent binge-drinking weekends with friends). He says that he does not smoke or use illicit drugs. Vital signs include: pulse rate 120/min, respiratory rate 26/min, and blood pressure 100/70 mm Hg. On examination, the patient’s abdomen is diffusely tender. His breath smells like alcohol, with a fruity tinge to it. Bowel sounds are present. No other findings are noted. Fingerstick glucose is 76mg/dL. After the examination, the patient suddenly and spontaneously vomits. Which of the following is the underlying mechanism of the most likely diagnosis in this patient?
A 34-year-old male visits the clinic with complaints of intermittent diarrhea over the past 6 months. He has lost 6.8 kg (15 lb) over that time period. His frequent bowel movements are affecting his social life and he would like definitive treatment. Past medical history is significant for chronic type 2 diabetes that is well controlled with insulin. No other family member has a similar condition. He does not smoke tobacco and drinks alcohol only on weekends. Today, his vitals are within normal limits. On physical exam, he appears gaunt and anxious. His heart has a regular rate and rhythm and his lungs are clear to auscultation bilaterally. Additionally, the patient has a red-purple rash on his lower abdomen, groin, and the dorsum of both hands. The rash consists of pruritic annular lesions. He is referred to a dermatologist for core biopsy which is consistent with necrolytic migratory erythema. Further workup reveals a large hormone secreting mass in the tail of his pancreas. Which of the following is the action of the hormone that is in excess in this patient?
A 51-year-old gentleman presents with new onset bilateral paresthesias of his feet. He also admits that he has not been able to exercise as much as previously and his friends have commented that he looks pale. Upon physical exam you find that he has conjunctival pallor and mildly decreased sensation and proprioception on his feet bilaterally. Based on your suspicions you decide to obtain a blood smear where you see megaloblasts as well as hypersegmented neutrophils. Given these findings you decide to investigate the cause of his disorder by injecting an intramuscular vitamin, then feeding him a radiolabeled version of the same vitamin orally. After waiting 24 hours you see that no radiolabeled vitamin appears in the urine so you repeat the test with intrinsic factor added to the oral mixture, at which point 20% of the radiolabeled vitamin appears in the urine. Which of the following is the most likely etiology of this gentleman's symptoms?
Practice by Chapter
Fatty acid oxidation (beta-oxidation)
Practice Questions
Fatty acid synthesis
Practice Questions
Ketone body metabolism
Practice Questions
Cholesterol synthesis and regulation
Practice Questions
Lipoprotein metabolism
Practice Questions
Phospholipid metabolism
Practice Questions
Eicosanoid synthesis and function
Practice Questions
Steroid hormone synthesis
Practice Questions
Adipose tissue metabolism
Practice Questions
Brown vs. white adipose tissue
Practice Questions
Disorders of lipid metabolism
Practice Questions
Integration with carbohydrate metabolism
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app