
Lipid metabolism — MCQs

On this page
A newborn female is found to have ambiguous genitalia and hypotension. Laboratory workup reveals hyperkalemia, hyperreninemia, and elevated levels of 17-hydroxyprogesterone in the patient's urine. Which of the following enzymes would you expect to be deficient in this patient?
An 8-month-old boy is brought to the physician by his parents for gradually increasing loss of neck control and inability to roll over for the past 2 months. During this time, he has had multiple episodes of unresponsiveness with a blank stare and fluttering of the eyelids. His parents state that he sometimes does not turn when called but gets startled by loud noises. He does not maintain eye contact. He was able to roll over from front to back at 5 months of age and has not yet begun to sit or crawl. His parents are of Ashkenazi Jewish descent. Neurological examination shows generalized hypotonia. Deep tendon reflexes are 3+ bilaterally. Plantar reflex shows extensor response bilaterally. Fundoscopy shows bright red macular spots bilaterally. The remainder of the examination shows no abnormalities. Which of the following is the most likely cause of this patient's symptoms?
A 22-year-old man comes to the physician because of a fall associated with a 6-month history of increasing difficulty walking. Over the last year, his friends have also noticed his speech becoming slower. During this period, he also gave up his hobby of playing video games because he has become clumsy with his hands. His father died of esophageal varices at the age of 40 years. The patient does not smoke or drink alcohol. He takes no medications. He appears sad. His temperature is 37°C (98.6°F), pulse is 70/min, and blood pressure is 120/80 mm Hg. He is alert and oriented to person, place, and time. His speech is slurred and monotonous; his gait is unsteady. Examination shows scleral icterus and some drooling. The liver is palpated 2 to 3 cm below the right costal margin, and the spleen is palpated 1 to 2 cm below the left costal margin. Further evaluation of this patient is most likely to show which of the following findings?
A medical student is spending his research year studying the physiology of cholesterol transport within the body. Specifically, he wants to examine how high-density lipoprotein (HDL) particles are able to give other lipoproteins the ability to hydrolyse triglycerides into free fatty acids. He labels all the proteins on HDL particles with a tracer dye and finds that some of them are transferred onto very-low-density lipoprotein (VLDL) particles after the 2 are incubated together. Furthermore, he finds that only VLDL particles with transferred proteins are able to catalyze triglyceride hydrolysis. Which of the following components were most likely transferred from HDL to VLDL particles to enable this reaction?
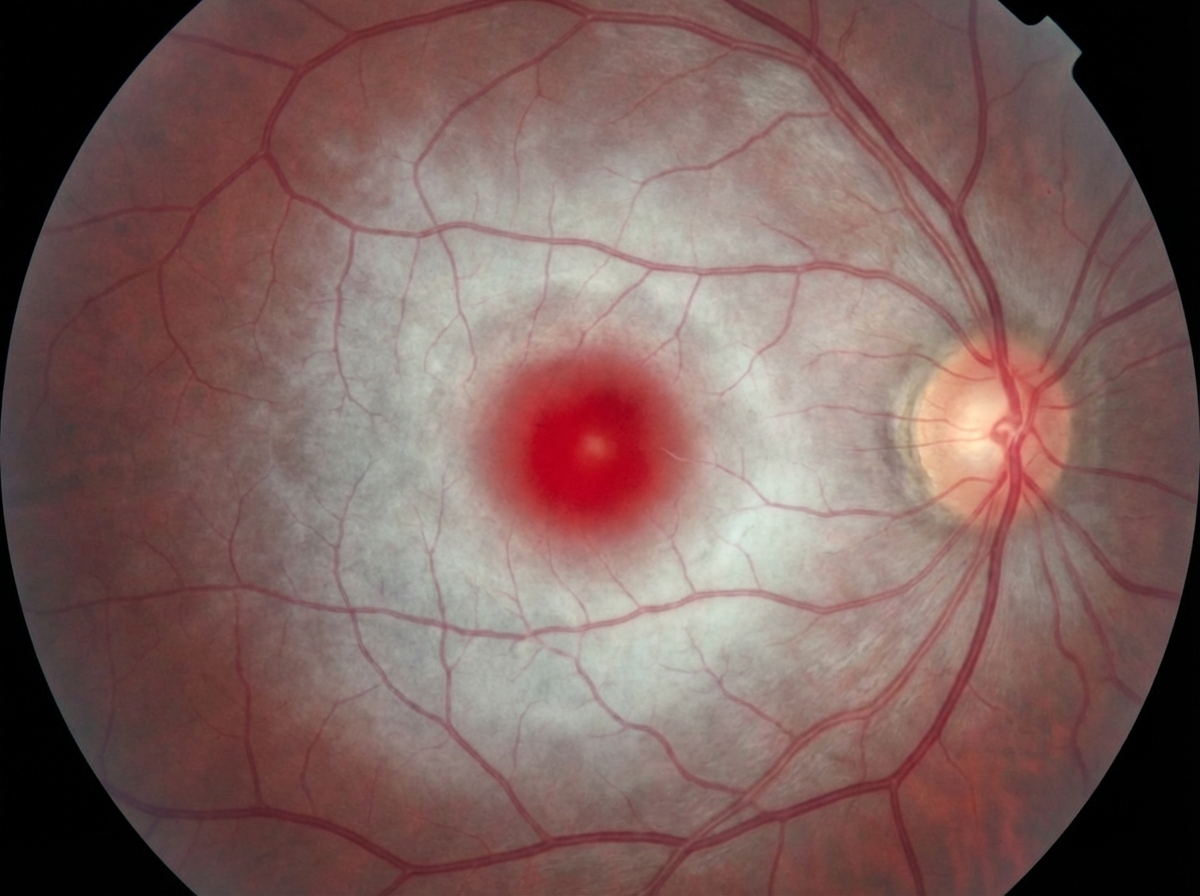
A 7-month-old boy is brought by his parents to the pediatrician’s office. His mother says the child has been weakening progressively and is not as active as he used to be when he was born. His condition seems to be getting worse, especially over the last month. He was born at 41 weeks through normal vaginal delivery. There were no complications observed during the prenatal period. He was progressing well over the 1st few months and achieving the appropriate milestones. On examination, his abdomen appears soft with no liver enlargement. The patient appears to be dehydrated and lethargic. The results of a fundoscopic examination are shown in the picture. A blood test for which of the following enzymes is the next best assay to evaluate this patient's health?
A 10-year-old boy is brought to the emergency department due to vomiting and weakness. He is attending a summer camp and was on a hike with the other kids and a camp counselor. His friends say that the boy skipped breakfast, and the counselor says he forgot to pack snacks for the kids during the hike. The child’s parents are contacted and report that the child has been completely healthy since birth. They also say there is an uncle who would have to eat regularly or he would have similar symptoms. At the hospital, his heart rate is 90/min, respiratory rate is 17/min, blood pressure is 110/65 mm Hg, and temperature is 37.0°C (98.6°F). Physical examination reveals a visibly lethargic child with slight disorientation to time and place. Mild hepatosplenomegaly is observed but no signs of dehydration are noted. A blood sample is drawn, and fluids are started via an intravenous line. Lab report Serum glucose 44 mg/dL Serum ketones absent Serum creatinine 1.0 mg/dL Blood urea nitrogen 32 mg/dL Alanine aminotransferase (ALT) 425 U/L Aspartate aminotransferase (AST) 372 U/L Hemoglobin (Hb%) 12.5 g/dL Mean corpuscular volume (MCV) 80 fl Reticulocyte count 1% Erythrocyte count 5.1 million/mm3 Which of the following is most likely deficient in this patient?
A 32-year-old woman presents to her family physician with a long history of depression, irritability, and, more recently, personality changes. As her partner comments, she has stopped engaging in activities she used to enjoy like dancing, drumming lessons, and yoga. The patient denies changes in skin pigmentation and assures she keeps a balanced diet low in fat and carbohydrates. During the physical examination, jaundice and dark rings encircling the iris of the eye are noted, as well as hepatomegaly and gait disturbances. For a follow-up visit, the patient brings a battery of laboratory tests that includes a complete blood count showing normocytic normochromic anemia, a negative Coombs, normal iron levels, normal fasting glucose levels, elevated aminotransferases from the liver biochemical tests, bilirubin, and decreased serum ceruloplasmin levels. Antinuclear antibodies are negative. What is the most likely diagnosis?
A 40-year-old G1P0010 presents to the clinic with nausea and vomiting 8 weeks after a spontaneous abortion at 10 weeks gestation. She admits to heavy drinking (7–8 glasses of wine per day) for the last 20 years; however, after the pregnancy loss, she increased her drinking to 8–9 glasses per day. Hepatomegaly, right upper quadrant pain, and jaundice are noted on abdominal examination. The lungs are clear to auscultation with no abnormalities on chest X-ray. Liver function tests are obtained and a biopsy is performed. Which of the following findings is most likely to be true in her condition?
A 60-year-old man presents to the physician for a regular checkup. The patient has a history of osteoarthritis in his right knee and gastroesophageal reflux disease. His conditions are well controlled by medications, and he has no active complaints at the moment. He takes ibuprofen, omeprazole, and a multivitamin. Laboratory tests show: Laboratory test Serum glucose (fasting) 77 mg/dL Serum electrolytes Sodium 142 mEq/L Potassium 3.9 mEq/L Chloride 101 mEq/L Serum creatinine 0.8 mg/dL Blood urea nitrogen 10 mg/dL Cholesterol, total 250 mg/dL HDL-cholesterol 35 mg/dL LDL-cholesterol 190 mg/dL Triglycerides 135 mg/dL Which of the following will be increased in the liver?
A 12-month-old child passed away after suffering from craniofacial abnormalities, neurologic dysfunction, and hepatomegaly. Analysis of the child’s blood plasma shows an increase in very long chain fatty acids. The cellular analysis demonstrates dysfunction of an organelle responsible for the breakdown of these fatty acids within the cell. Postmortem, the child is diagnosed with Zellweger syndrome. The family is informed about the autosomal recessive inheritance pattern of the disease and their carrier status. Which of the following processes is deficient in the dysfunctional organelle in this disease?
Practice by Chapter
Fatty acid oxidation (beta-oxidation)
Practice Questions
Fatty acid synthesis
Practice Questions
Ketone body metabolism
Practice Questions
Cholesterol synthesis and regulation
Practice Questions
Lipoprotein metabolism
Practice Questions
Phospholipid metabolism
Practice Questions
Eicosanoid synthesis and function
Practice Questions
Steroid hormone synthesis
Practice Questions
Adipose tissue metabolism
Practice Questions
Brown vs. white adipose tissue
Practice Questions
Disorders of lipid metabolism
Practice Questions
Integration with carbohydrate metabolism
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app