
Lipid metabolism — MCQs

On this page
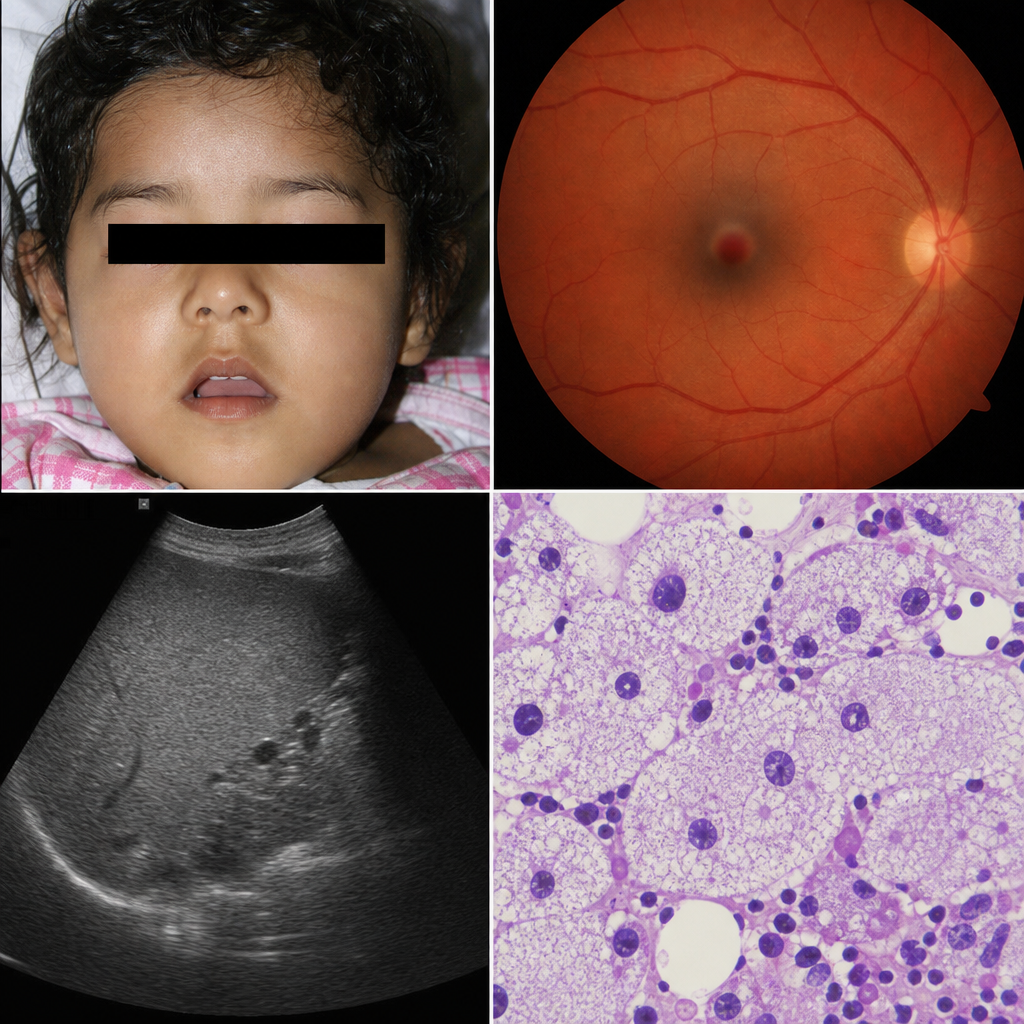
A 3-year-old girl presents with progressive neurodegeneration, a cherry-red spot on fundoscopic examination, and hepatosplenomegaly. Bone marrow biopsy reveals large macrophages with foamy cytoplasm. Enzyme assay of leukocytes shows markedly reduced activity of a lysosomal hydrolase. Which of the following correctly identifies the deficient enzyme and its accumulating substrate in this condition?
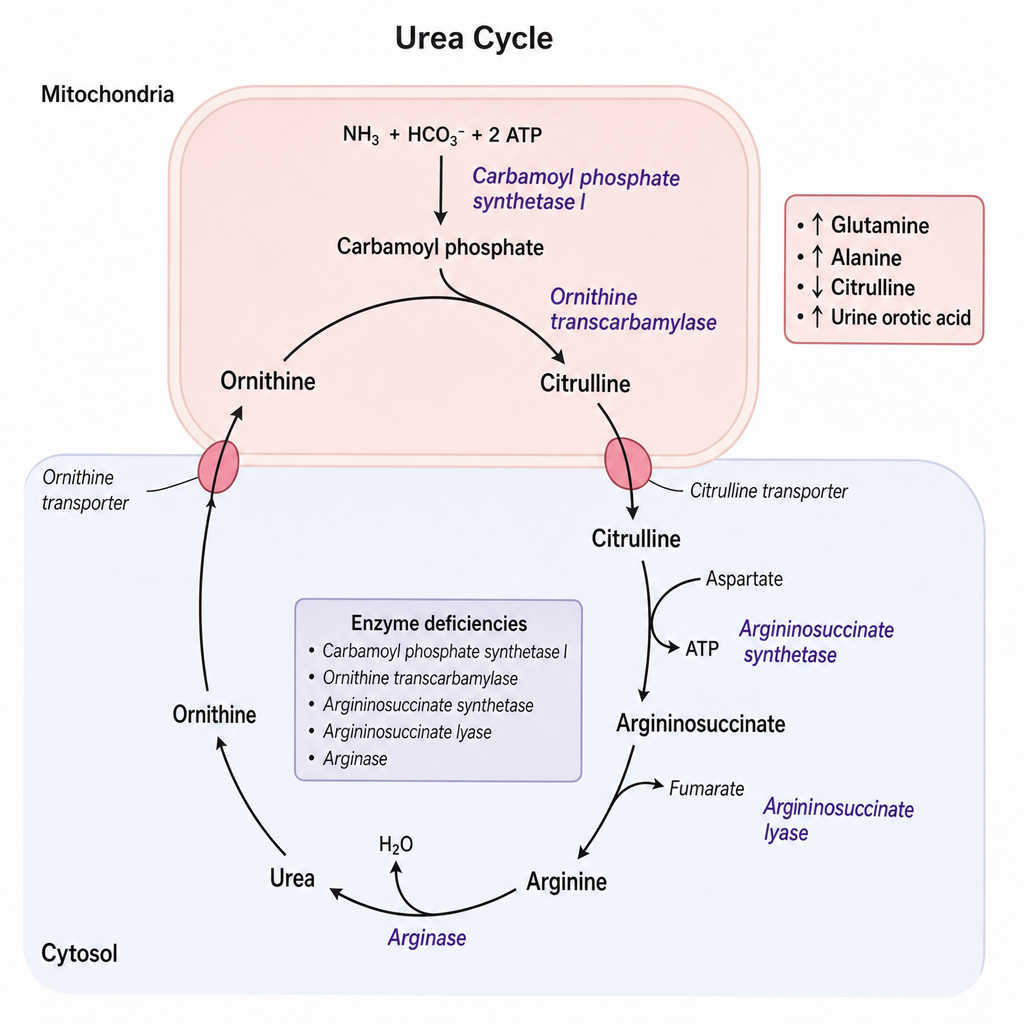
A 3-year-old girl is evaluated for recurrent episodes of vomiting, lethargy, and hyperammonemia that worsen after high-protein meals. Plasma amino acid analysis reveals elevated glutamine and alanine with low citrulline. Urine orotic acid is markedly elevated. Her older brother died at age 2 years with a similar presentation. She is a symptomatic heterozygous female due to skewed X-inactivation. The condition follows an X-linked recessive inheritance pattern. Based on the biochemical findings and the inheritance pattern, which enzyme is most likely deficient in this patient?
Colipase is an enzyme found in secretions from which of the following? (Recent NEET Pattern 2018)
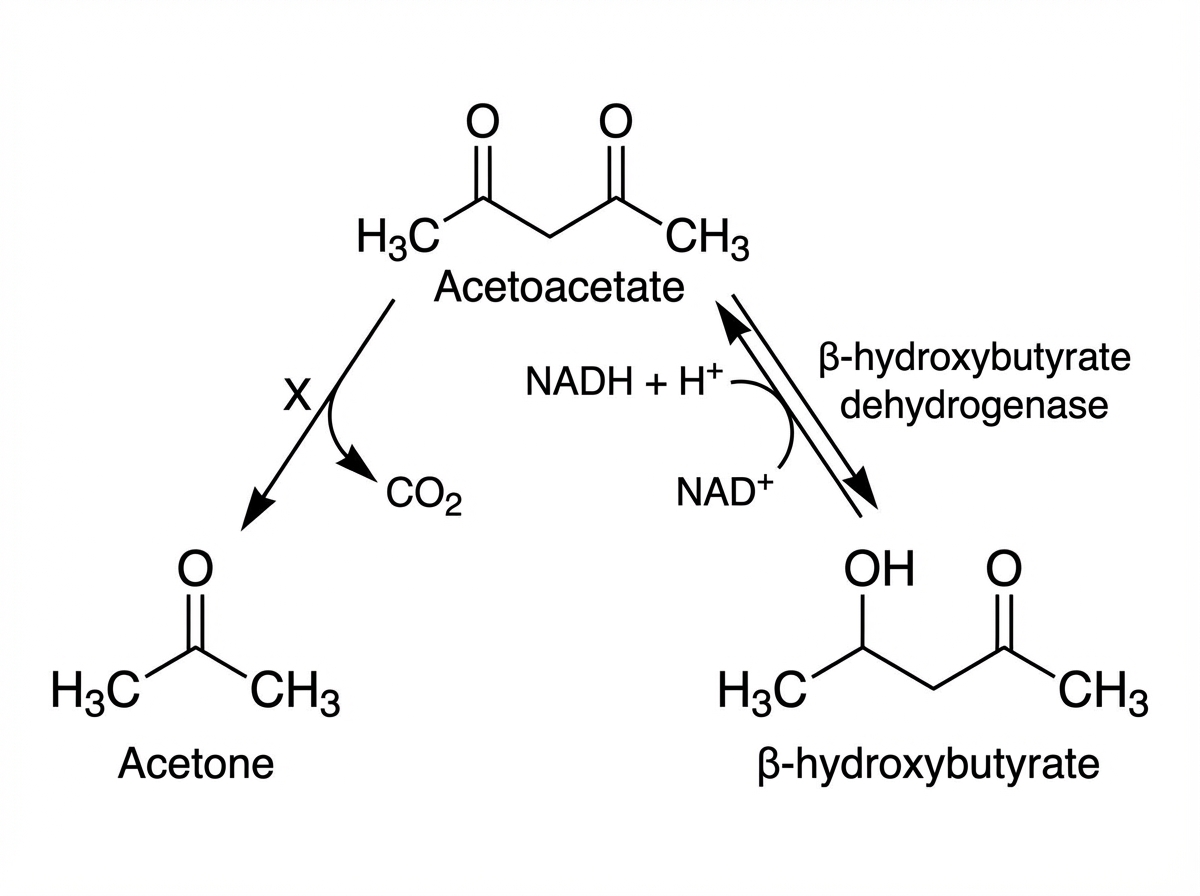
Name the mechanism shown as $X$ below.
Which is the location of HMG CoA synthase in cholesterol metabolism?
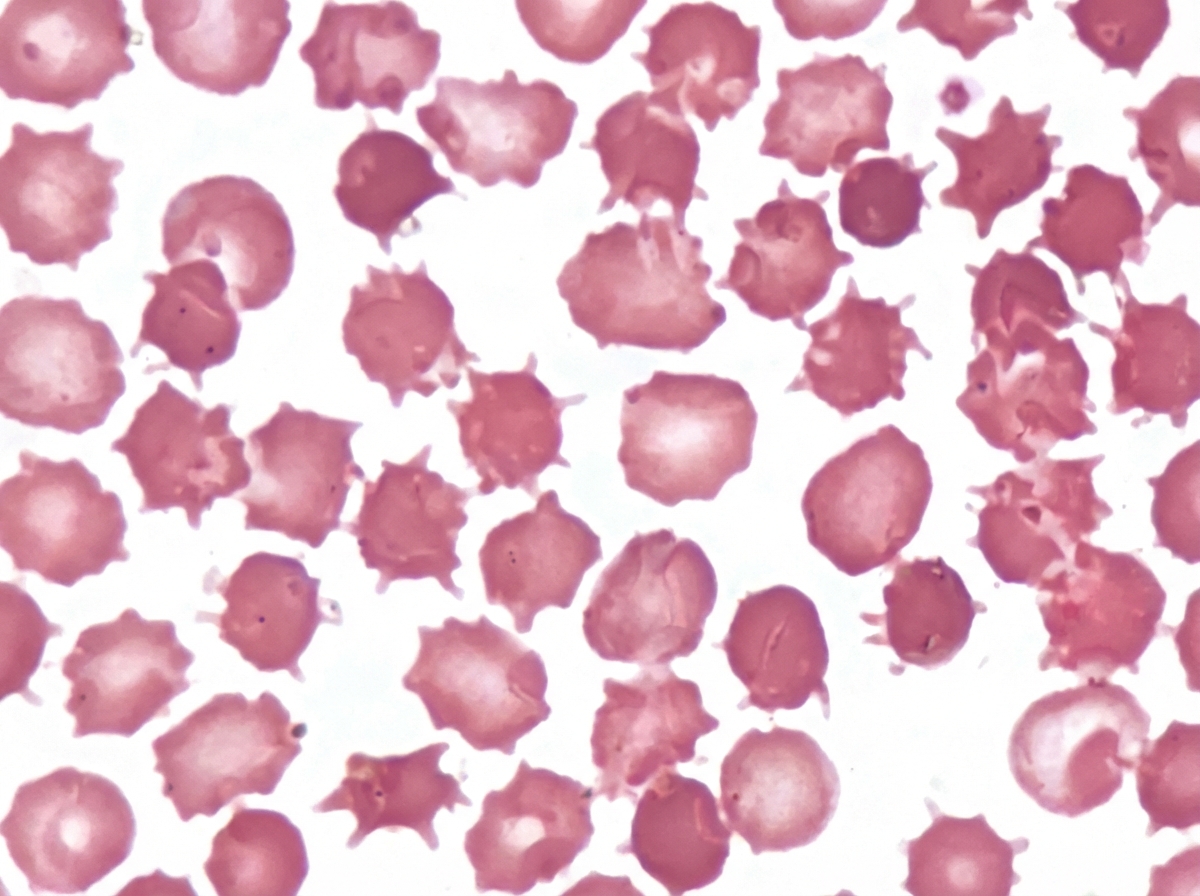
An adult male presented with a protruding abdomen, diarrhea, visual symptoms, and neurological manifestations. His LDL is low. Based on the peripheral smear finding shown in the image, what is the likely diagnosis?
A patient came to the hospital with severe abdominal pain, and lipase levels were elevated. On imaging, a stone is found in the common bile duct (CBD). Which enzyme is most likely elevated in this condition?
A patient came to the emergency room with severe abdominal pain. The serum triglyceride level was $1500 \mathrm{mg} / \mathrm{dL}$. What is the most likely defect?
A patient has multiple tendon xanthomas. Serum cholesterol ( $398 \mathrm{mg} / \mathrm{dL}$ ) and LDL ( 220 $\mathrm{mg} / \mathrm{dL}$ ) were found to be elevated. What is the most likely defect?
A patient presents with xanthomas on the Achilles tendon. What is the most likely diagnosis?
Practice by Chapter
Fatty acid oxidation (beta-oxidation)
Practice Questions
Fatty acid synthesis
Practice Questions
Ketone body metabolism
Practice Questions
Cholesterol synthesis and regulation
Practice Questions
Lipoprotein metabolism
Practice Questions
Phospholipid metabolism
Practice Questions
Eicosanoid synthesis and function
Practice Questions
Steroid hormone synthesis
Practice Questions
Adipose tissue metabolism
Practice Questions
Brown vs. white adipose tissue
Practice Questions
Disorders of lipid metabolism
Practice Questions
Integration with carbohydrate metabolism
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app