
Glycolysis — MCQs

On this page
A 4-year-old boy is brought to the pediatrician because his parents noticed he has difficulty climbing stairs and rising from the floor. He also has a history of recurrent respiratory infections and his parents report he snores loudly at night. On examination, he has mild hepatomegaly and cardiomegaly is noted on chest X-ray. Serum creatine kinase is markedly elevated. Muscle biopsy shows large vacuoles within myocytes that stain intensely with periodic acid-Schiff (PAS). Electron microscopy reveals membrane-bound organelles packed with glycogen. Which enzyme deficiency is responsible for this presentation?
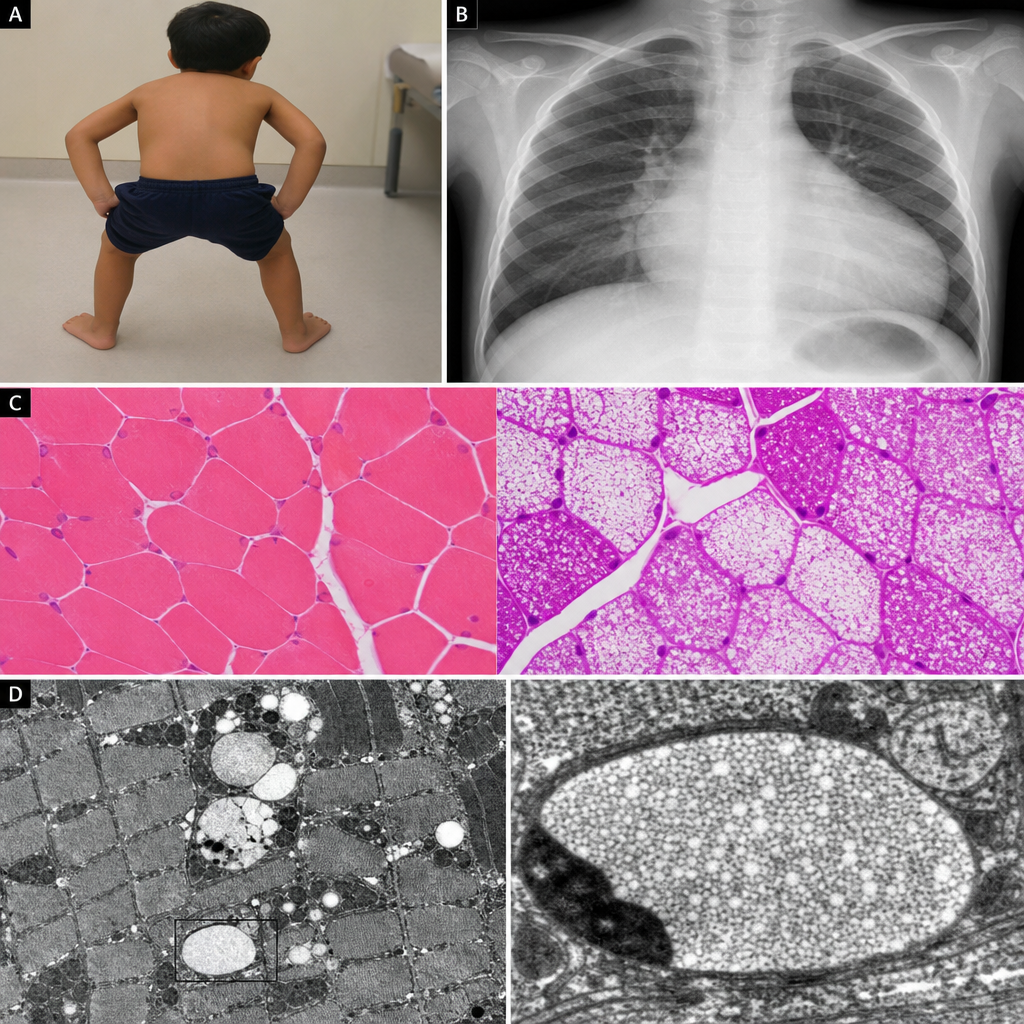
A 3-month-old boy is brought to the pediatrician because his parents are concerned about poor feeding, difficulty breathing, and marked floppiness since birth. On examination, he has profound hypotonia, macroglossia, and significant cardiomegaly on chest X-ray. He requires supplemental oxygen due to respiratory distress. Serum creatine kinase is markedly elevated. A muscle biopsy is obtained and sent for electron microscopy, which demonstrates large membrane-bound organelles packed with electron-dense granular material distending the muscle fibers, with near-complete absence of normal myofibrillar architecture in affected regions. Which of the following enzyme deficiencies is most consistent with these findings?
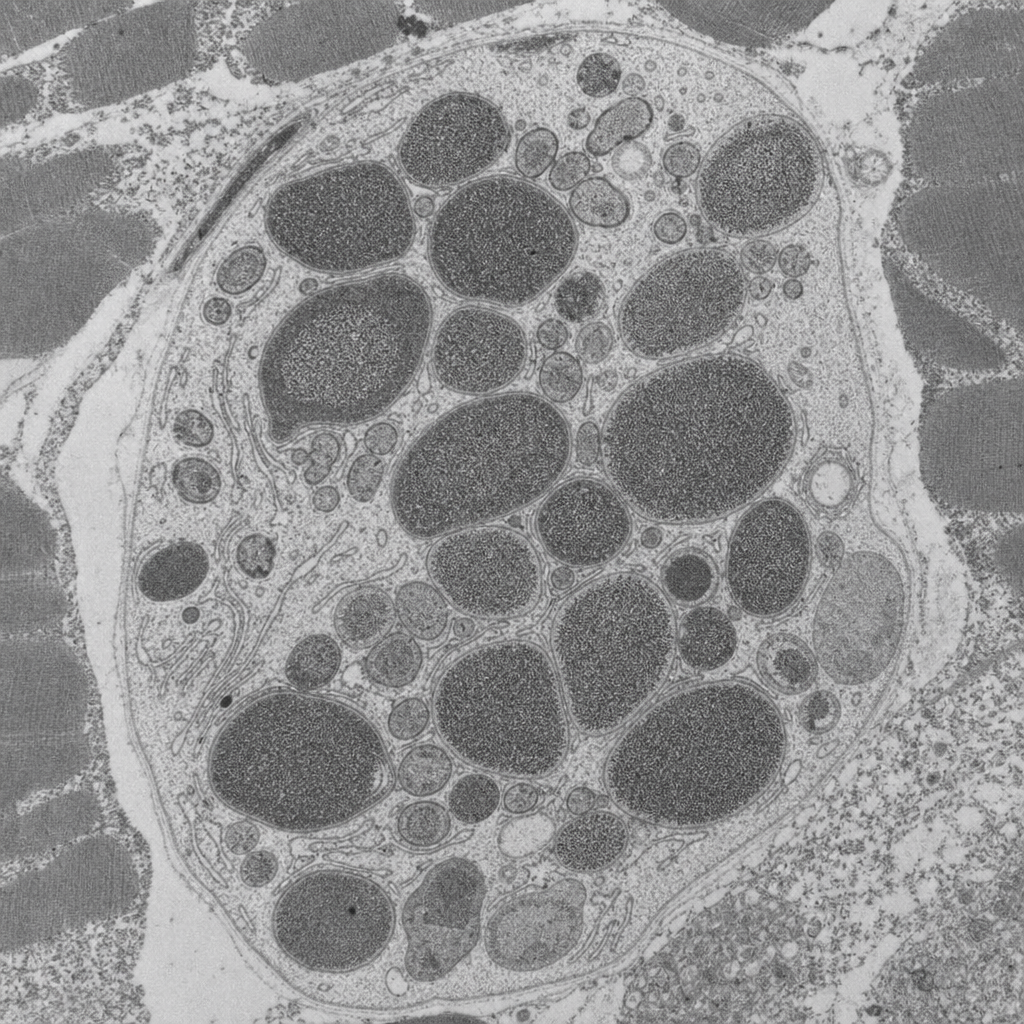
A 16-year-old teenager is brought to the emergency department after having slipped on ice while walking to school. She hit her head on the side of the pavement and retained consciousness. She was brought to the closest ER within an hour of the incident. The ER physician sends her immediately to get a CT scan and also orders routine blood work. The physician understands that in cases of stress, such as in this patient, the concentration of certain hormones will be increased, while others will be decreased. Considering allosteric regulation by hormones, which of the following enzymes will most likely be inhibited in this patient?
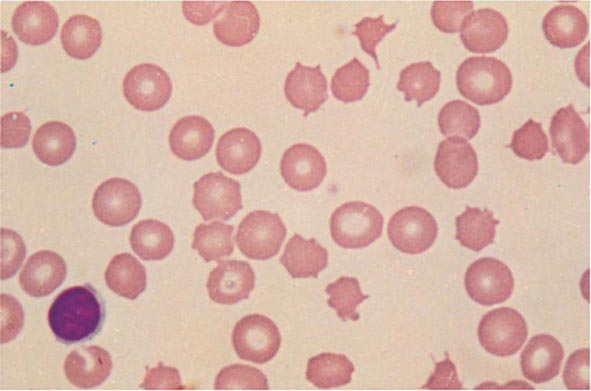
A mother brings her newborn baby to the pediatrician after noting that his skin looks yellow. The patient's lactate dehydrogenase is elevated and haptoglobin is decreased. A smear of the child's blood is shown below. The patient is ultimately found to have decreased ability to process phosphoenolpyruvate to pyruvate. Which of the following metabolic changes is most likely to occur in this patient?
Researchers are investigating a new mouse model of glycogen regulation. They add hepatocyte enzyme extracts to radiolabeled glucose to investigate glycogen synthesis, in particular two enzymes. They notice that the first enzyme adds a radiolabeled glucose to the end of a long strand of radiolabeled glucose. The second enzyme then appears to rearrange the glycogen structure such that there appears to be shorter strands that are linked. Which of the following pairs of enzymes in humans is most similar to the enzymes being investigated by the scientists?
Maturity Onset Diabetes of the Young (MODY) type 2 is a consequence of a defective pancreatic enzyme, which normally acts as a glucose sensor, resulting in a mild hyperglycemia. The hyperglycemia is especially exacerbated during pregnancy. Which of the following pathways is controlled by this enzyme?
To maintain blood glucose levels even after glycogen stores have been depleted, the body, mainly the liver, is able to synthesize glucose in a process called gluconeogenesis. Which of the following reactions of gluconeogenesis requires an enzyme different from glycolysis?
A 22-year-old man presents to his primary care provider because of fever, diarrhea, and abdominal cramps. He has returned from Dhaka, Bangladesh recently where he was visiting his relatives. He is diagnosed with Shigella infection, and ciprofloxacin is started. He develops severe nausea and weakness 2 days later and complains of passing dark urine. The lab test results reveal a hemoglobin level of 7.9 g/dL, increased unconjugated bilirubin, increased reticulocyte count, increased lactate dehydrogenase, and increased blood urea. Which of the following is the best next step for the diagnosis of this patient’s condition?
To prepare for an endoscopy, a 27-year-old male was asked by the gastroenterologist to fast overnight for his 12 p.m. appointment the next day. Therefore, his last meal was dinner at 5 p.m. the day before the appointment. By 12 p.m. the day of the appointment, his primary source of glucose was being generated from gluconeogenesis, which occurs via the reversal of glycolysis with extra enzymes to bypass the irreversible steps in glycolysis. Which of the following irreversible steps of gluconeogenesis occurs in the mitochondria?
A 2-year-old boy presents to the emergency department with new onset seizures. After controlling the seizures with fosphenytoin loading, a history is obtained that reveals mild hypotonia and developmental delay since birth. There is also a history of a genetic biochemical disorder on the maternal side but the family does not know the name of the disease. Physical exam is unrevealing and initial lab testing shows a pH of 7.34 with a pCO2 of 31 (normal range 35-45) and a bicarbonate level of 17 mEq/L (normal range 22-28). Further bloodwork shows an accumulation of alanine and pyruvate. A deficiency in which of the following enzymes is most likely responsible for this patient's clinical syndrome?
Practice by Chapter
Overview and regulation of glycolysis
Practice Questions
Preparatory phase reactions
Practice Questions
Pay-off phase reactions
Practice Questions
Rate-limiting enzymes and control points
Practice Questions
Energy yield and ATP production
Practice Questions
Fates of pyruvate
Practice Questions
Substrate-level phosphorylation
Practice Questions
Feeder pathways to glycolysis
Practice Questions
Glycolysis in different tissues
Practice Questions
Disorders of glycolytic enzymes
Practice Questions
Alternative glycolytic pathways
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app