
Glycogen storage diseases — MCQs

On this page
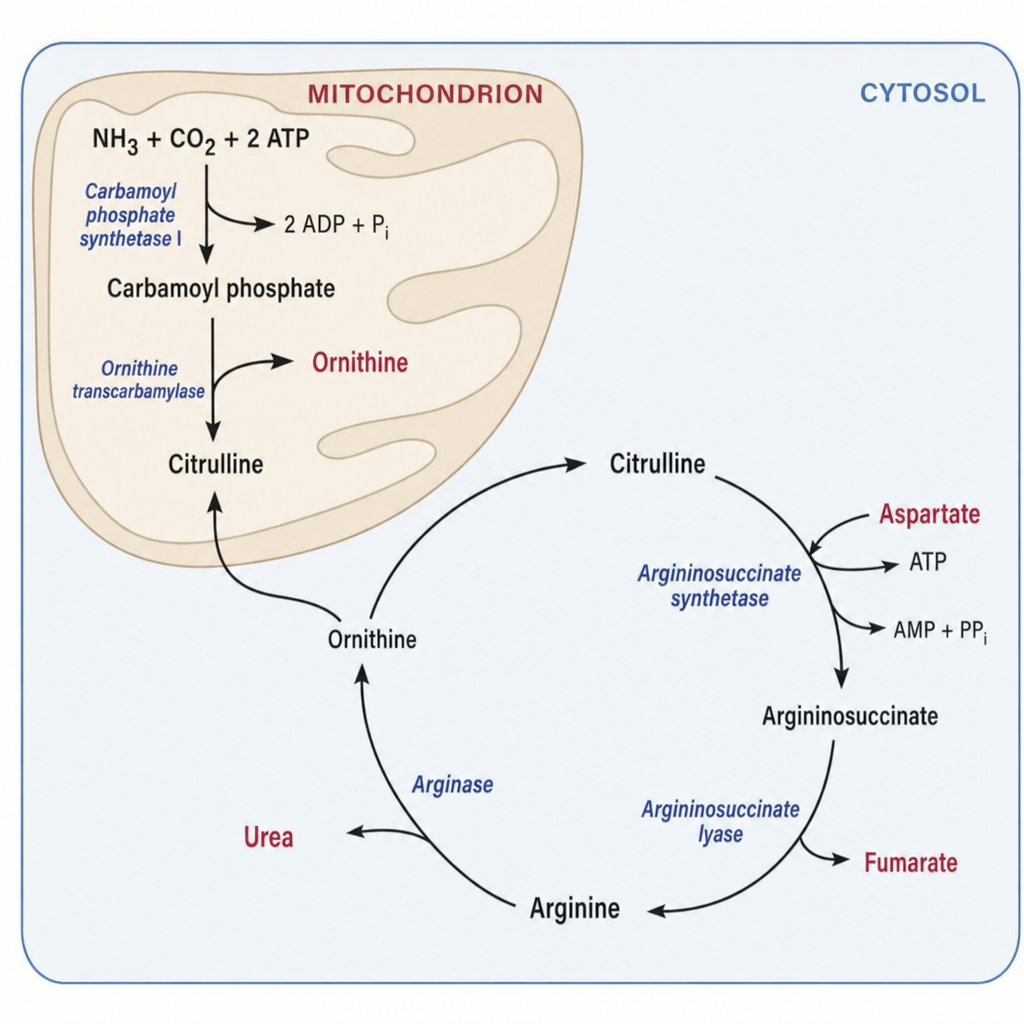
A 3-day-old male neonate born to consanguineous parents develops lethargy, poor feeding, and seizures. Laboratory results show hyperammonemia (NH3 = 380 µmol/L), elevated plasma citrulline, and markedly elevated plasma argininosuccinate. Urine organic acid analysis shows argininosuccinic aciduria. The enzymatic block in this patient prevents which of the following conversions in the urea cycle?
A pharmaceutical company is developing a novel therapy for Type I glycogen storage disease using adeno-associated virus (AAV) vectors to deliver the glucose-6-phosphatase gene. Phase I trials show successful hepatic gene transfer with 30% of normal enzyme activity, resulting in improved fasting glucose and reduced hepatomegaly. However, some patients develop immune responses to the viral vector and lose therapeutic benefit after 6 months. Synthesize the therapeutic approach and evaluate strategies to optimize long-term efficacy.
A 25-year-old woman with Type I glycogen storage disease has been managed with frequent feedings and nocturnal gastric drip feeding since childhood. She now presents for preconception counseling. Her current labs show: fasting glucose 65 mg/dL, lactate 4 mmol/L, uric acid 8 mg/dL, triglycerides 400 mg/dL, and ALT 150 U/L. Renal ultrasound shows bilateral adenomas. She desires pregnancy but is concerned about risks. Synthesize the multisystem complications and evaluate the pregnancy management approach.
A neonate presents with severe hypotonia, feeding difficulties, and respiratory distress requiring mechanical ventilation. Echocardiography reveals severe hypertrophic cardiomyopathy with left ventricular outflow obstruction. Genetic testing confirms Pompe disease, and enzyme replacement therapy (ERT) with recombinant alpha-glucosidase is initiated. At 6 months, the infant shows improved motor function and reduced cardiomegaly, but cognitive development remains delayed. The parents question the differential response. Evaluate the pathophysiological basis for this treatment response pattern.
A 15-year-old athlete undergoes genetic testing after experiencing exercise-induced myoglobinuria. Testing reveals a homozygous mutation in the PYGM gene. His younger brother, age 12, is found to be heterozygous for the same mutation and is asymptomatic. The parents request guidance on the younger brother's athletic participation. Analyze the genotype-phenotype correlation and evaluate the appropriate counseling.
A 5-year-old boy with known glycogen storage disease presents to the emergency department with altered mental status. He missed his nighttime cornstarch feeding due to viral gastroenteritis with vomiting. Labs show: glucose 35 mg/dL, lactate 8 mmol/L (elevated), uric acid 12 mg/dL (elevated), triglycerides 600 mg/dL (elevated), and mild metabolic acidosis. His abdomen is distended with massive hepatomegaly. Analyze the metabolic derangements and determine the priority management.
A 2-year-old boy presents with progressive hepatosplenomegaly and failure to thrive. Liver biopsy shows accumulation of abnormal glycogen with long outer branches and fewer branch points. The child develops portal hypertension and ascites. Laboratory evaluation reveals hypoglycemia is not prominent, but liver function tests are markedly abnormal with elevated transaminases and bilirubin. Analyze these findings to determine the most likely diagnosis and underlying pathophysiology.
A 3-year-old boy presents with hepatomegaly, growth retardation, and fasting hypoglycemia that is less severe than his sibling who has Von Gierke disease. Laboratory studies show elevated transaminases and mild hyperlipidemia, but normal lactate and uric acid levels. Unlike his sibling, he responds to glucagon administration with increased blood glucose when fed, but not after prolonged fasting. Apply these findings to identify the enzymatic defect.
A 4-year-old girl presents with progressive muscle weakness, hypotonia, and cardiomegaly. Chest X-ray shows massive cardiomegaly with pulmonary congestion. Echocardiography reveals severe left ventricular hypertrophy with outflow obstruction. Electromyography shows myopathic changes, and serum creatine kinase is markedly elevated. Muscle biopsy shows vacuoles filled with glycogen and increased lysosomal acid phosphatase activity. Apply this information to determine the deficient enzyme.
A 20-year-old man presents with muscle cramps and myoglobinuria after moderate exercise. He reports that he gets a 'second wind' if he rests briefly during exercise and then continues. Ischemic forearm exercise test shows no rise in venous lactate. Muscle biopsy reveals periodic acid-Schiff (PAS)-positive material. Apply these findings to identify the underlying enzyme deficiency.
Practice by Chapter
Glycogen structure and metabolism overview
Practice Questions
GSD type I (von Gierke disease)
Practice Questions
GSD type II (Pompe disease)
Practice Questions
GSD type III (Cori disease)
Practice Questions
GSD type IV (Andersen disease)
Practice Questions
GSD type V (McArdle disease)
Practice Questions
GSD type VI (Hers disease)
Practice Questions
GSD type VII (Tarui disease)
Practice Questions
Clinical manifestations by tissue involvement
Practice Questions
Diagnostic approaches to GSDs
Practice Questions
Management strategies for GSDs
Practice Questions
Genetics and inheritance of GSDs
Practice Questions
Want unlimited practice?
Get full access to all questions, explanations, and performance tracking.
Scan to download app